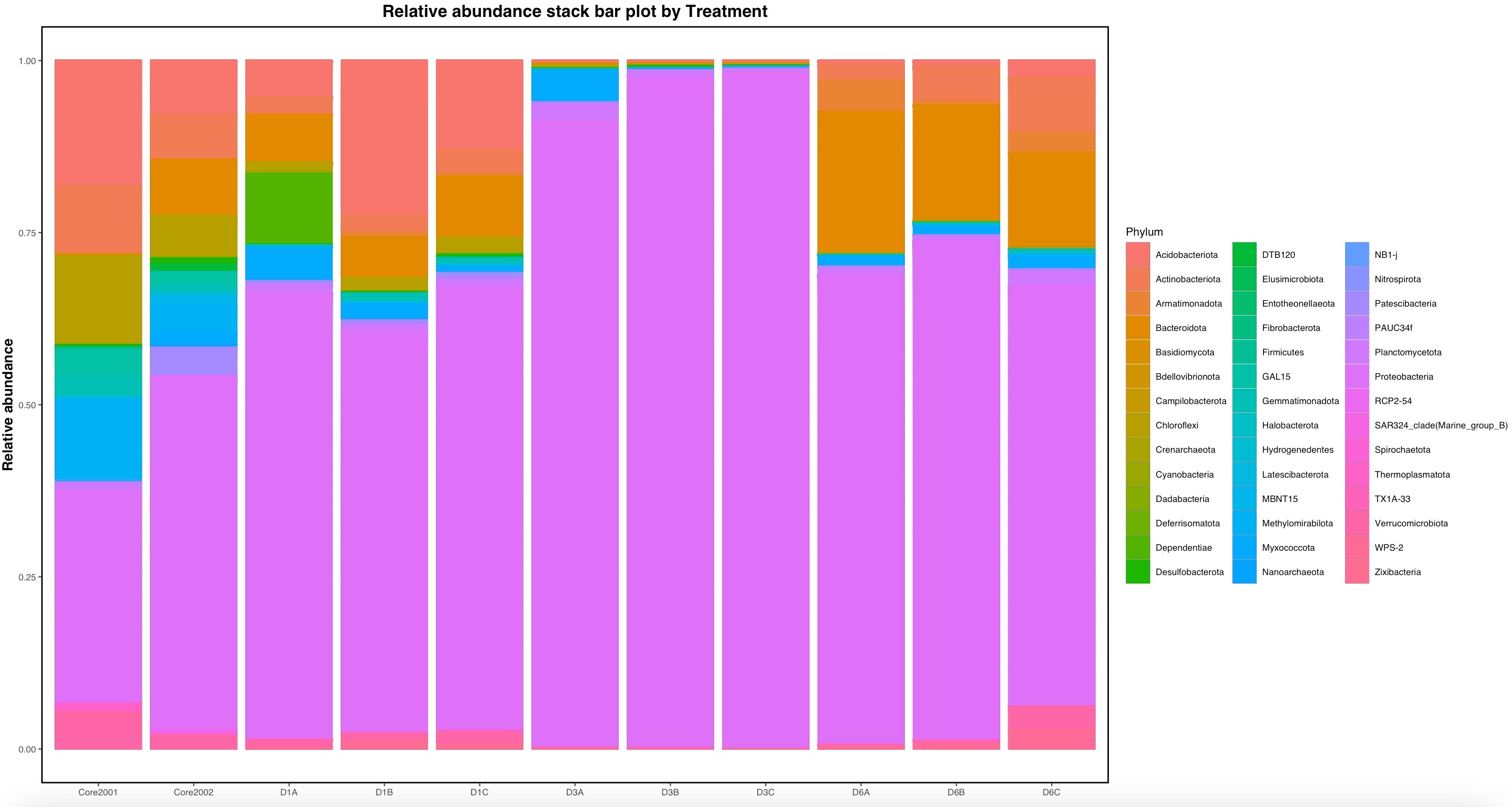
I am working on an environmental microbiome project, studying bacterial communities cultured from sediment core near an oil spill in Bemidji, Minnesota.
I would like to use qiime2 artifacts from my data set to produce a stacked relative abundance bar chart by phylum. Except I would like one phylum to be further divided into smaller taxa. Specifically, I would like to divide my Proteobacteria phylum divided in to alpha, beta, gamma, and delta Proteobacteria on the bar chart. Attached are my preliminary results.
I would also like to change the color of each bacteria type, so the neighboring bacteria on the chart are a starkly different color, rather than being represented by an indistinguishable gradient.
I figured it might be helpful if I included the code I used to produce the attached rel abundance stack bar plot in my previous post:
library(phyloseq)
###tools>install packages>devtools #devtools::install_github("jbisanz/qiime2R") #BiocManager::install("Rtools")
library(qiime2R)
library(tidyverse)
library(vegan)
library(picante)
library(cowplot)
library(DESeq2)
library(ggplot2)
###tools>install packages>picante
###tools>install packages>cowplot
###tools>install packages>ggplot2 #BiocManager::install("DESeq2")
###change working directory>more>go to working directory> r studio
###manually move all files (in next command) to r studio
phy <- qza_to_phyloseq (features = "DOC_ASV_taxo_contaminants_removed-table.qza", "DOC-taxonomy.qza", tree = "DOC-rooted-tree.qza", metadata = "DOC_map_contaminants_removed.tsv")
###manually move file shannon_vector.qza to r studio
###to see new file created "phy" go to environment tab
shannon <- read_qza("shannon_vector.qza")
rich <- plot_richness(phy)
###highlight only rich and press command enter
nsamples(phy)
sample_names(phy)
OTUtable<-otu_table(phy)
OTUtable<-as.data.frame(OTUtable)
write.csv(OTUtable,"OTU_table.csv")
rich
###pick up at line 199 in tutorial###
phy_rel_abund <- transform_sample_counts(phy, function(x) {x / sum(x)})
plot_bar(phy_rel_abund, fill = "Phylum") +
geom_bar(aes(color = Phylum, fill = Phylum), stat = "identity", position = "stack") +
labs(x = "", y="Relative abundance") +
theme_q2r() +
ggtitle("Relative abundance stack bar plot by Treatment") +
theme(axis.title = element_text(color="black", face="bold", size=10)) +
theme(plot.title = element_text(color="black", face = "bold", size =12, hjust = 0.5))