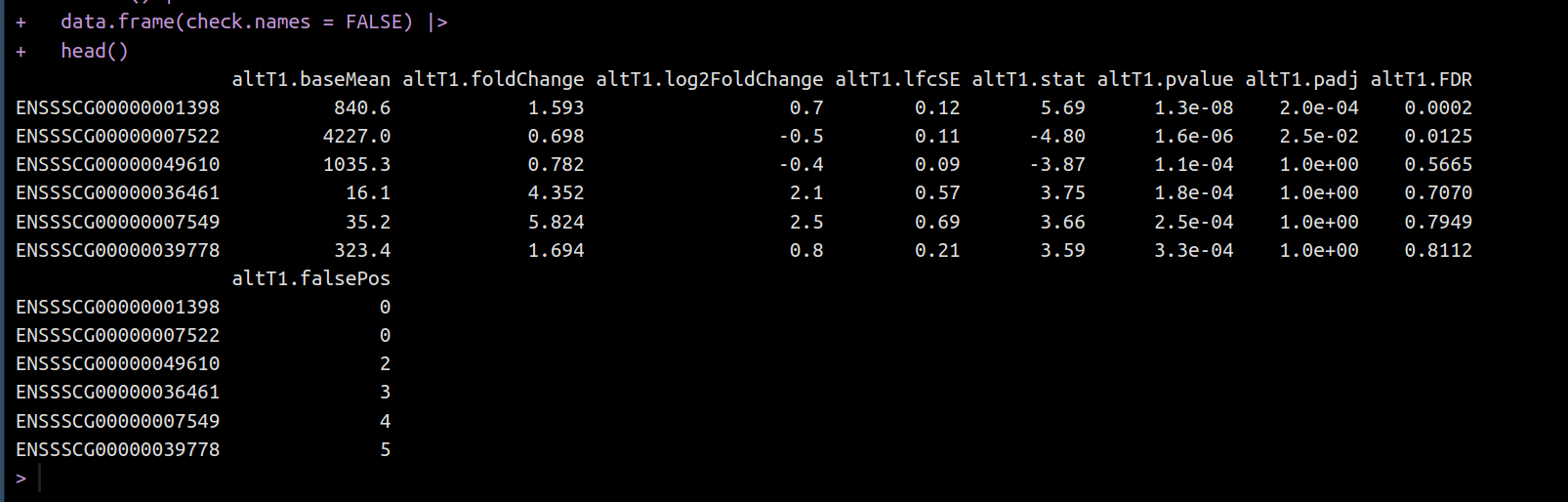
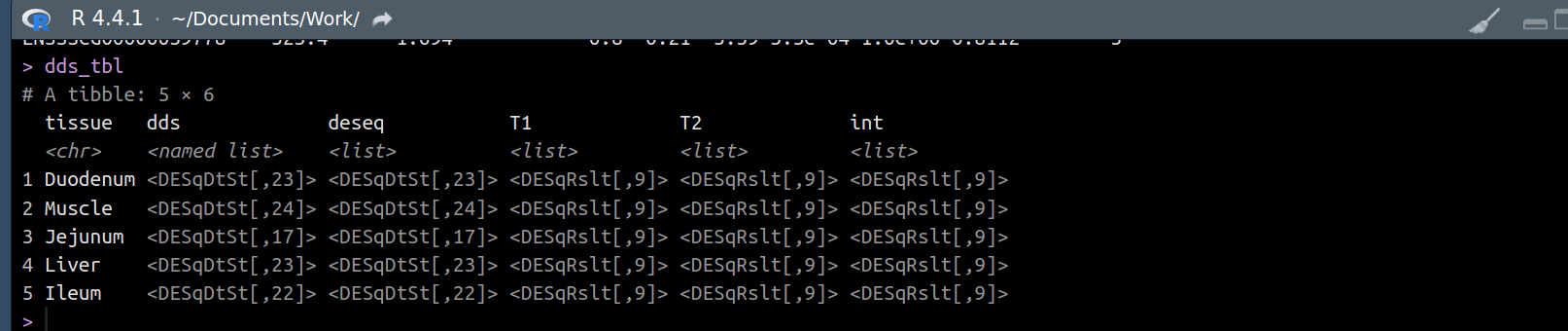
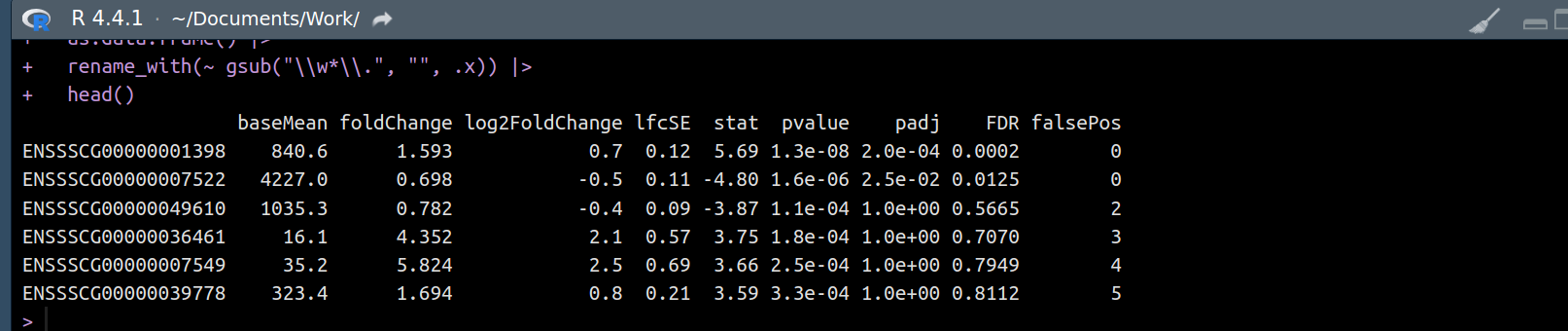
Hi all,
This is a difficult one for me to create a sample data set so I will try to describe as clearly as possible.
Basically, I want to access the data inside nested lists, not just the names of the data.
I have a list object called res_list. It contains 5 list objects (1 for each of 5 tissues). Each of these list objects contains 3 DESeqResults class objects. So a total of 15 deseqresults objects stored.
res_list[[1]][1] # extracts the first deseqresults object in the first tissue
res_list[[1]][2] # extracts the second deseqresults object in the first tissue
res_list[[1]][3] # extracts the third deseqresults object in the first tissue
res_list[[2]][1] # extracts the first deseqresults object in the second tissue
res_list[[2]][2] # and so on...
...
res_list[[5]][3] # extracts the final deseqresults object in the final tissue
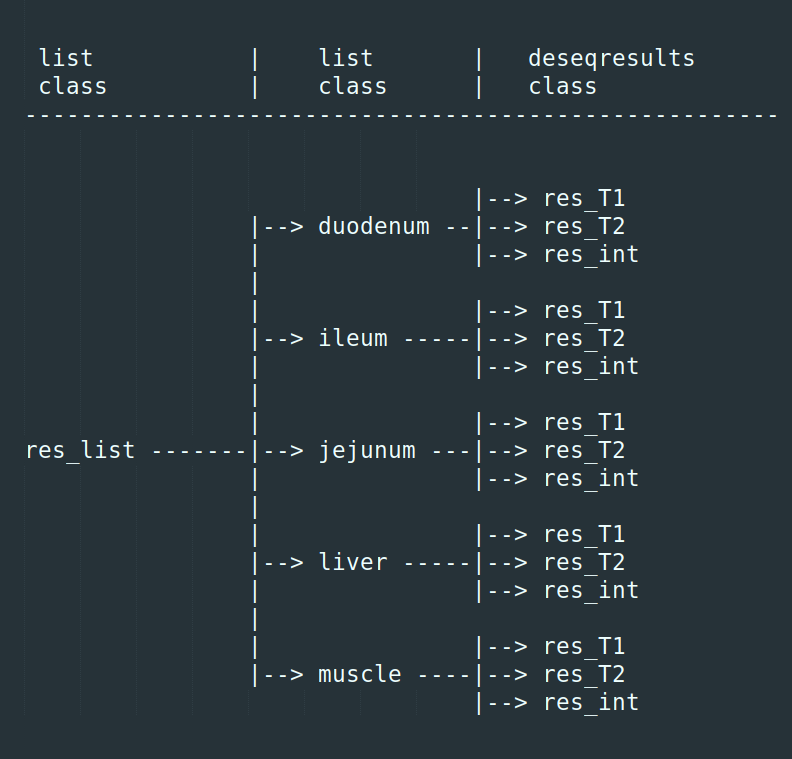
I need to transform each of the 15 deseq2results objects (adding/rearranging columns, filtering/ordering rows etc) But, I am struggling to access them. I have thought of a for loop in a for loop, an lapply in an lapply, and so on.
There is quite alot of transformation to do so I don't want to spend days coding myself into a dead end. I will be storing the resulting transformed data frames in the same way in a new list object
My best attempt so far has been:
for (tissue in names(res_list)) {
x <- names(test_list[[tissue]][1:3])
#x <- names(test_list[[tissue]][[1:3]]) # Error in test_list[[tissue]][[1:3]] : recursive indexing failed at level 2
lapply(x,function(x){
# res_T1
if (x == "res_T1"){
print(x)
}
# res_T2
# res_int
})
}
returns:
[1] "res_T1"
[1] "res_T1"
[1] "res_T1"
[1] "res_T1"
[1] "res_T1"
So what I can see is that I am just accessing the names of each object, but not the the object itself.
So print(x) returns prints "res_T1" 5 times (1 for each tissue), instead of printing the 5 deseq2results objects called "res_T1". If I can't even print it, I certainly cant transform it.
If I try the following instead of print(x), I just get a concatenated stream:
print(paste(res_list,tissue, x, sep="$") # i.e. res_list$Duodenum$res_T1
Another idea is to use a incremental counter to increase res_list[[ x ]][ y ] and process one at a time but Im not even sure if Ill hit the same prob.
I hate nested lists, but it seemed like the most efficient way of storing all this data in 1 place, and it's what I have now sooooo....
Thanks in advance.
Kenneth