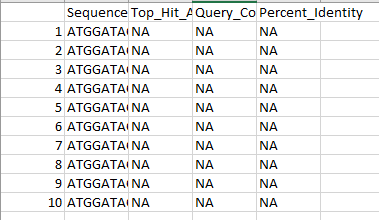
I'm trying to write an RStudio script that takes an excel file with a list of DNA sequences and searches BLASTn from NCBI with them, obtains the top hit for percent identity and accession number, and exports the results as a csv. The problem I am having is that the results are only showing NA for each DNA sequence in the new csv file instead of providing the expected results. I've attached the file that BLAST has been giving me with the NA values. Here is my code:
Read Excel file with DNA sequences
excel_file <- "C:/Users/rbh6/Documents/BLAST TEST START/TEST_BLAST_RSTUDIO.xlsx"
dna_sequences <- readxl::read_excel(excel_file, sheet = "Sheet1", col_names = TRUE)
Print the structure of dna_sequences to understand its format
print(head(dna_sequences))
Read Excel file with DNA sequences
excel_file <- "C:/Users/rbh6/Documents/BLAST TEST START/TEST_BLAST_RSTUDIO.xlsx"
dna_sequences <- readxl::read_excel(excel_file, sheet = "Sheet1", col_names = TRUE)
Print the structure of dna_sequences to understand its format
print(head(dna_sequences))
Perform BLAST search for each sequence
blast_results <- lapply(dna_sequences$DNA Sequence, function(sequence) {
result <- tryCatch({
perform_blast(sequence)
}, error = function(e) {
cat("Error in BLAST search for sequence:", sequence, "\n")
cat("Error message:", conditionMessage(e), "\n")
return(rep(NA, 3)) # Return NAs for columns if no match is found
})
if (length(result) == 0) {
return(rep(NA, 3))
} else {
return(result)
}
})
Determine the length of the longest vector among blast_results
max_length <- max(sapply(blast_results, function(x) max(lengths(x))))
Create a data frame with specified lengths, filling with NA where needed
result_df <- data.frame(
Sequence = rep(dna_sequences$DNA Sequence, each = max_length),
Top_Hit_Accession_Number = unlist(lapply(blast_results, function(x) c(x[[1]], rep(NA, max_length - length(x[[1]]))))),
Query_Coverage = unlist(lapply(blast_results, function(x) c(x[[2]], rep(NA, max_length - length(x[[2]]))))),
Percent_Identity = unlist(lapply(blast_results, function(x) c(x[[3]], rep(NA, max_length - length(x[[3]])))))
)
Export the data frame as a CSV file
setwd("C:/Users/rbh6/Documents/BLAST TEST COMPLETED")
data_Path <- "C:/Users/rbh6/Documents/BLAST TEST COMPLETED"
path <- file.path(data_Path, paste(Sys.Date(), "_BLAST_RESULTS.csv", sep = ""))
write.csv(result_df, file = path)
Display a message indicating successful completion
cat("BLAST search and CSV export completed. Results saved in", path, "\n")
cat("BLAST search completed. Displaying results:\n")
print(blast_results)