rnaseqDRaMA - RNAseq data visualization and mining
Authors: Yurii Chinenov, Max Chao, David Oliver
Working with Shiny more than 1 year
Abstract: RNAseq has been widely adopted as the method of choice for large-scale gene expression profiling. Data under-utilization, however remains a major challenge due to specific skill set required for data processing, interpretation, and analysis. To simplify end-user RNA-seq data interpretation, we created RNA-seq DRaMA (RNAseq Data Retrieval and Mining Analytical platform) - an R/Shiny interactive reporting system with user-friendly web interface for data exploration and visualization (https://hssgenomics.shinyapps.io/RNAseq_DRaMA/). The app supports many methods for data exploration including: sample PCA and multidimensional scaling, gene- and sample- correlation analyses, Venn diagram and UpSet set visualizations, gene expression group barplots and heatmaps with hierarchical clustering, volcano plots, pathway analysis with QuSAGE, and Transcription Factor network analysis. All plots are highly customized in terms of sample, feature, threshold, and color selections and create publication-ready pdf and tabular outputs. All features are well-documented with an in-app manual. RNAseq DRaMA has been extensively tested at the HSS Genomics Center with more than 100 projects delivered and several projects currently deployed in the public domain
Full Description:

In design of rnaseqDRaMA we were guided by several goal:
• Compatibility with existing RNA-seq data processing pipelines
• Interactivity via web-based interface
• Consistent user interface
• Customizable graphic and tabular outputs
RNA-seq DRaMA does not allow the end user to perform initial differential expression analysis (DEA), but rather provides an interface to explore the analysis performed by a bioinformatician. Although implementing Shiny-based DEA is technically possible we decided not to provide this capability at a very early design stage.
Three factors contributed to this decision:
• Computationally many aspects of creating the input for the platform are intractable for a typical user’s hardware (e.g. pathway analysis).
• There is always a balance between accessibility and power, that is, a platform which could also perform DEA would inherently be more complex, and as a result, would be accessible to fewer users (especially to high-level decision-makers that have limited time to learn the technical nuances of DEA). From the very beginning we hoped to create a platform with a shallow learning curve.
• It is almost impossible to foresee specific experimental designs or the need for an unusual model to account for a variety of experimental and nuisance variables. Experimental requirements often necessitate creating complex contrasts that cannot be easily automated. As such a platform with DEA would lack generality and would require constant tweaking in response to new, unique experimental requirements.
The RNA-seq DRaMA app relies on R Shiny, shinydashboard, plotly and ggplot2 packages to implement the framework for interactive data visualization. Several additional packages were used to support specific tasks. All methods for RNA-seq data exploration and visualization are accessible from links on the sidebar. Each method is extensively parameterized to customize analyses and graphic outputs. Internal clipboard functionality allows genes selection from one analysis to be used as input for a different method. Limited wildcard support allows gene selection based on patterns (eg. IL* will select all genes that start with “IL”). Plots generated by the app can be saved in pdf or png formats. The app is extensively documented: brief description of each plot/method is provided in-app in the description boxes, more detailed description of functionality is available via Manual at the sidebar. Currently, rnaseqDRaMA can be run as a local app in R environment or as a web application hosted on a shiny server or public services such as shinyapps.io. The app supports many methods for RNAseq data exploration, which we grouped into sections listed below:
Summary - Provides a brief experiment summary including a link to sequencing quality control and statistics provided, effective number of reads for each sample, a table of samples and experimental variables, a gene-wise variance plot, and a P-value histogram.
PCA - Principal component analysis (PCA) and classic multidimensional scaling analysis (MDS) of samples provides methods of reducing the number of variables describing your system to a few dimensions (called principal components, PC) that describe the largest sources of variation in your system. In RNA-seq, and other sequencing technologies, PCA is an efficient visualization tool for quickly identifying treatment effects on gene expression. It is also useful for diagnosing possible technical issues such as poor replicate reproducibility. PCA loadings heatmaps helps to determine principal component with largest contribution to a specific experimental variable
Correlation - This section contains a sample correlation plot and gene coexpression heatmap to quickly identify outlying samples, visualize pairwise similarities between samples, and identify groups of genes whose expression changes in the same direction across all samples. R heatmaply package was used to create interactive heatmaps in rnaseqDRaMA.
Set Intersections - Venn Diagrams and their more exquisite counterpart, Upset Plots, provide an overview of overlaps between differentially expressed genes in different treatment/condition combinations. This section relies on several extensively modified functions from the VennDiagram package and UpSetR package
Heatmap - The Highly Variable Gene Heatmap is useful for identification of genes which are strongly altered across samples/conditions. In addition if a large enough number of samples are collected, heatmaps can inform the presence of outliers and genes which are most important for the observed effect of a treatment. The Custom Gene Selection Heatmap accepts gene names from a user and plots their expression changes across conditions (logCPMs) or comparisons (logFC). Combined with sets of genes from Set Intersections, custom heatmaps will give additional insight into how gene regulation is changing between conditions.
Gene Expression - This section provides access to the table of raw and normalized read counts for each expressed gene identified in the experiment and creates customized bar graphs for user selected gene sets.
Differential Expression - Visualizes the results of differential expression analysis in the form of a volcano plot with several levels of gene selection. A Volcano Plot combines a measure of statistical significance (P-Value or FDR) with the magnitude of the change between compared samples.
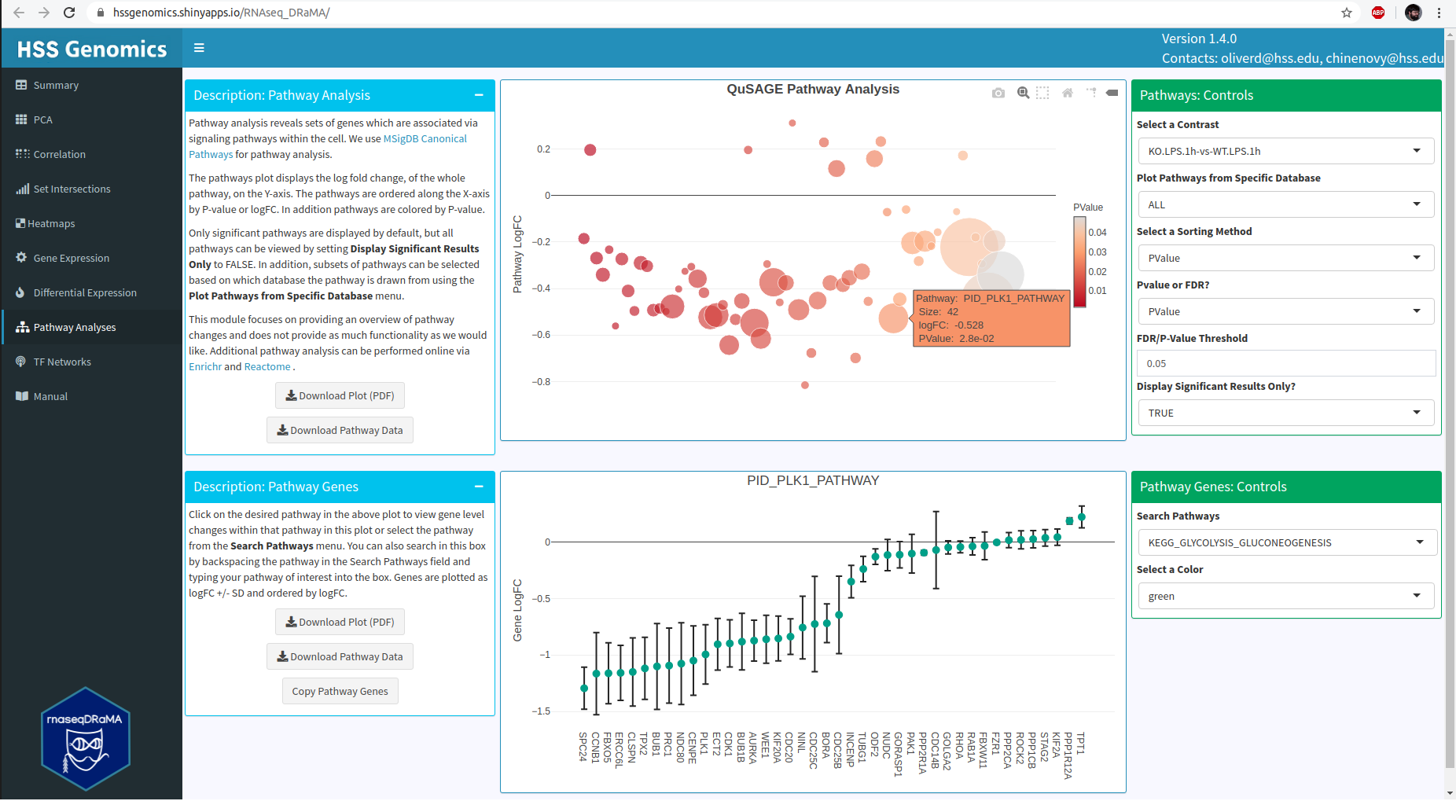
Pathway Analysis - Visualizes the results of QuSAGE pathway analysis that reveals functionally-related sets of genes that are potentially co-regulated, and therefore may affect the outcome of an entire signaling or metabolic pathway.
TF Networks - Transcription Factor Networks section identifies transcription factors in a supplied set of genes and links them to their targets within the same set of genes producing a network graph representation. If only a single transcription factor gene is selected, a network of nearest neighbors of that TF will be shown. These networks are built based on AnimalTFDB 3.0 and RegNetwork which includes both transcription factors and transcriptional co-factors.
Category: Research
Keywords: Bioinformatics, RNAseq, gene expression, data visualization and mining
Shiny app: https://hssgenomics.shinyapps.io/RNAseq_DRaMA/
Repo: hssgenomics / Shiny · GitLab
RStudio Cloud: Posit Cloud
Thumbnail:

Full image: