0
I am trying out GLMMs models to test whether two categorical variables (species and sex) and their interaction (sex + species + sex*species= fixed factors) influence certain acoustic parameters (response variable) of some vocalisations. My response variables are continuous numerical positive, such as the Fundamental frequency or duration of vocalizations. So, I'm trying different glmms for every acoustic parameter I have. My model has the ID as random factor, and the context of emission of vocalizations as another fixed variable. For each individual I have more than one observation, meaning that each individual vocalized more than once.
My response variables don't have a normal distribution, some of them are very asymmetrical, and one of them has two central humps. I have tried for example with the fundamental frequency, I've done a logarithmic transformation and used gamma, gaussian or inverse.gaussian with various link functions (log, inverse, identity), but when I check the assumptions (normality and homogeneity of residuals) these are not verified.
The general model setting is:
full_F <- glmer(Fundam_freq ~ Sex + Breed + Sex*Breed +
Context + (1 | Cat_ID), data = data, family = ?(link = "?"))
EXAMPLE WITH family=Gamma(link = "log") after having log-transformed + 1 the response variable. Here I attach images of checking residuals assumptions ![]()
- Checking assumptions:
DHARMa::simulateResiduals(fittedModel = full_F, plot = T)
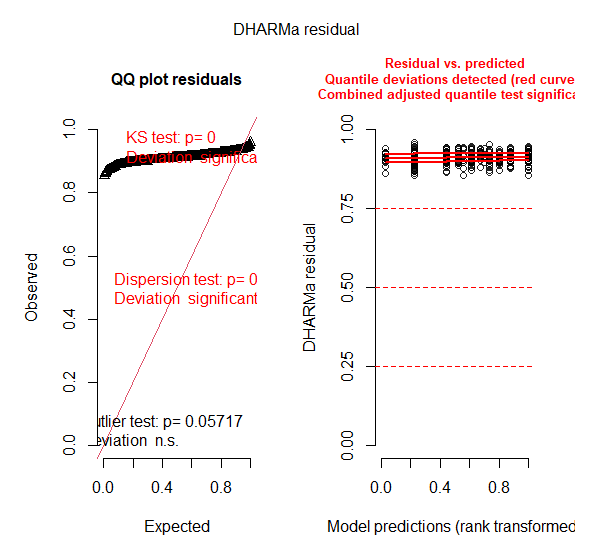
- Checking assumptions with:
sjPlot::plot_model(full_F, type = "diag")
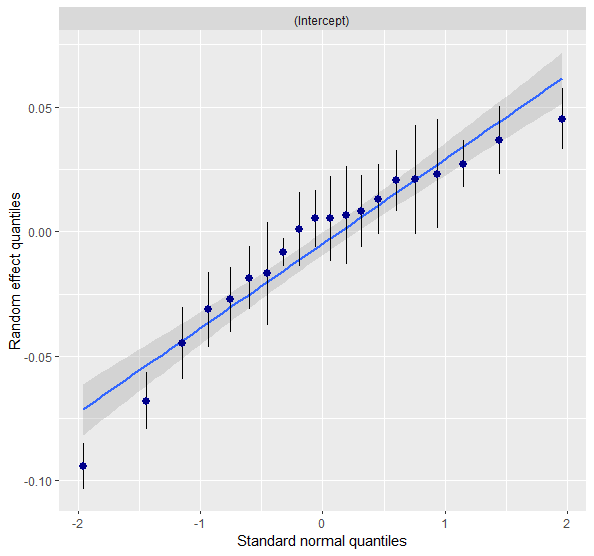
- Checking assumptions with:
diagnostics.plot(full_F)
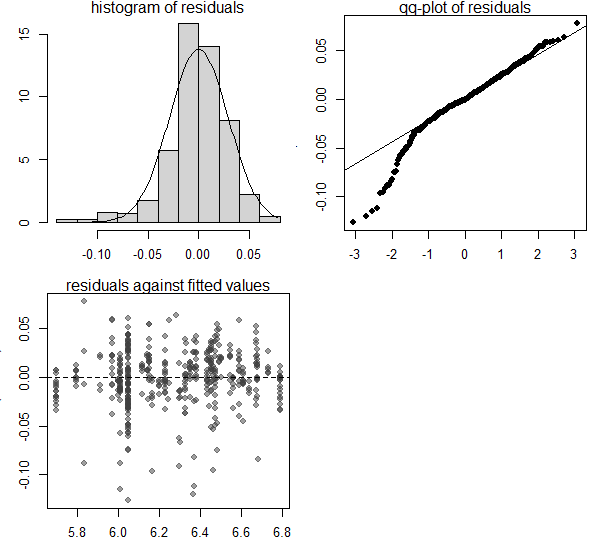
Which families and link functions do you recommend? Is there something I should do before run the glmm? (obviously, I transformed the Sex, Breed and Context into factors before run the model)
Thank you very much!