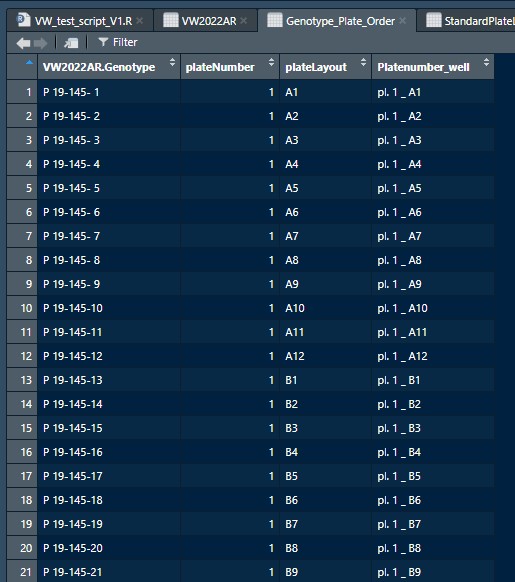
I have a list called Genotype_Plate_Order in which thousands of samples are listed. Each sample has a sample name (VW2022AR.Genotype; example P 19-145- 01) and a sample position (Platenumber_well).
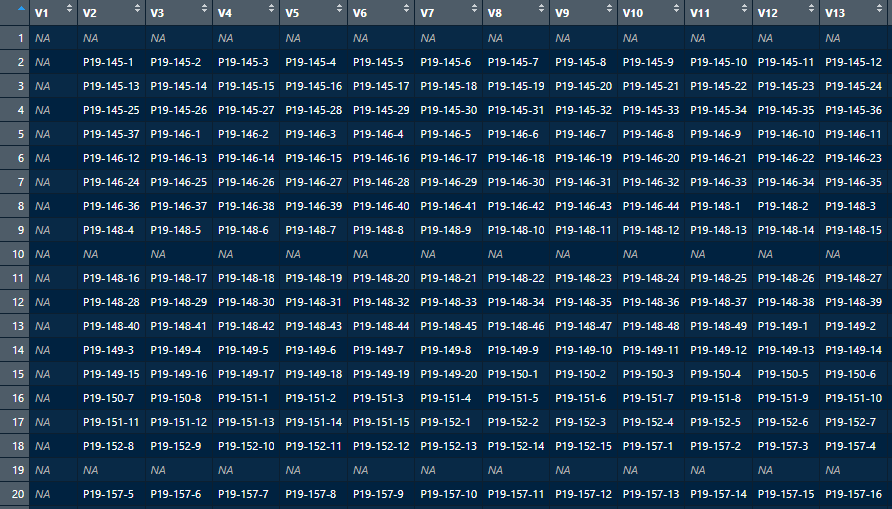
One plate holds 96 different wells, from A1 till H12. The combination of plate number and well number is unique. Since there are thousands of samples in this list, there are also many plate numbers of course.
Below are the first 6 rows of this dataframe.
VW2022AR.Genotype plateNumber plateLayout Platenumber_well
1 P 19-145- 1 1 A1 pl. 1 _ A1
2 P 19-145- 2 1 A2 pl. 1 _ A2
3 P 19-145- 3 1 A3 pl. 1 _ A3
4 P 19-145- 4 1 A4 pl. 1 _ A4
5 P 19-145- 5 1 A5 pl. 1 _ A5
6 P 19-145- 6 1 A6 pl. 1 _ A6
Top-view of this list with samples:
The vector "Platenumber_well" needs to be looked up in the dataframe 'StandardPlateLayout'. See a head() example and printscreen below.
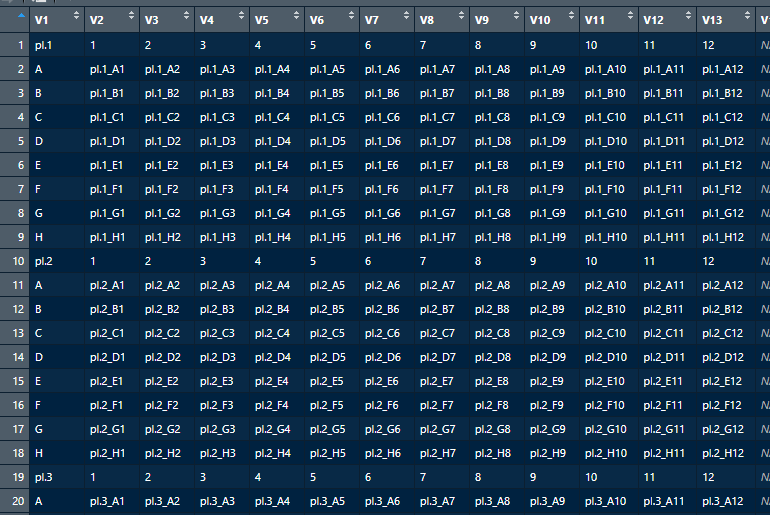
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13 V14 V15 V16 V17
1 pl.1 1 2 3 4 5 6 7 8 9 10 11 12 NA NA NA NA
2 A pl.1_A1 pl.1_A2 pl.1_A3 pl.1_A4 pl.1_A5 pl.1_A6 pl.1_A7 pl.1_A8 pl.1_A9 pl.1_A10 pl.1_A11 pl.1_A12 NA NA NA NA
3 B pl.1_B1 pl.1_B2 pl.1_B3 pl.1_B4 pl.1_B5 pl.1_B6 pl.1_B7 pl.1_B8 pl.1_B9 pl.1_B10 pl.1_B11 pl.1_B12 NA NA NA NA
4 C pl.1_C1 pl.1_C2 pl.1_C3 pl.1_C4 pl.1_C5 pl.1_C6 pl.1_C7 pl.1_C8 pl.1_C9 pl.1_C10 pl.1_C11 pl.1_C12 NA NA NA NA
5 D pl.1_D1 pl.1_D2 pl.1_D3 pl.1_D4 pl.1_D5 pl.1_D6 pl.1_D7 pl.1_D8 pl.1_D9 pl.1_D10 pl.1_D11 pl.1_D12 NA NA NA NA
6 E pl.1_E1 pl.1_E2 pl.1_E3 pl.1_E4 pl.1_E5 pl.1_E6 pl.1_E7 pl.1_E8 pl.1_E9 pl.1_E10 pl.1_E11 pl.1_E12 NA NA NA NA
7 F pl.1_F1 pl.1_F2 pl.1_F3 pl.1_F4 pl.1_F5 pl.1_F6 pl.1_F7 pl.1_F8 pl.1_F9 pl.1_F10 pl.1_F11 pl.1_F12 NA NA NA NA
8 G pl.1_G1 pl.1_G2 pl.1_G3 pl.1_G4 pl.1_G5 pl.1_G6 pl.1_G7 pl.1_G8 pl.1_G9 pl.1_G10 pl.1_G11 pl.1_G12 NA NA NA NA
9 H pl.1_H1 pl.1_H2 pl.1_H3 pl.1_H4 pl.1_H5 pl.1_H6 pl.1_H7 pl.1_H8 pl.1_H9 pl.1_H10 pl.1_H11 pl.1_H12 NA NA NA NA
10 pl.2 1 2 3 4 5 6 7 8 9 10 11 12 NA NA NA NA
11 A pl.2_A1 pl.2_A2 pl.2_A3 pl.2_A4 pl.2_A5 pl.2_A6 pl.2_A7 pl.2_A8 pl.2_A9 pl.2_A10 pl.2_A11 pl.2_A12 NA NA NA NA
12 B pl.2_B1 pl.2_B2 pl.2_B3 pl.2_B4 pl.2_B5 pl.2_B6 pl.2_B7 pl.2_B8 pl.2_B9 pl.2_B10 pl.2_B11 pl.2_B12 NA NA NA NA
This 'StandardPlateLayout' dataframe is a standard excel dataframe that explaines the order of samples in a collection box that is used for sample-collections. The dataframe is hundreds of plates (pl.##) long.
The objective is: The vector "Platenumber_well" from the dataframe 'Genotype_Plate_Order' needs to be looked up in the dataframe 'StandardPlateLayout'.
When true, it needs to be overwritten by the sample-name from the vector 'VW2022AR.Genotype' from 'Genotype_Plate_Order'.
I came up so far with the code below but somehow it gives me all NA. If someone has any suggestion, it would be very appreciated.
StandardPlateLayout[] <- lapply(
StandardPlateLayout, function(x) Genotype_Plate_Order$VW2022AR.Genotype[match(x,
Genotype_Plate_Order$Platenumber_well)])