Hello everyone, does anyone of you know what this error means? (Error in .local(object, ..., value): all(!is.na(value)) is not TRUE)
I had already done this analysis many times and I don't know what is happening now.
library(readxl)
Family <- read_excel("~/RSTUDIO/Datos_cianobacterias/ciano_otu_family_rdp.xlsx")
#NORMALIZATION OF RAW READ TABLE LOG2
#OTUS en filas, columnas son las muestras, es al reves que para el resto de pasos
data<- Family
replicates <- as.data.frame(colnames(data)[-1])
colnames(replicates) <- "replicates"
attach(Family)
rwnames <- OTU
data <- as.matrix(data[,-1])
rownames(data) <- rwnames
data[is.na(data)] <- 0
library(DESeq2)
#> Loading required package: S4Vectors
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#> Loading required package: parallel
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:parallel':
#>
#> clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
#> clusterExport, clusterMap, parApply, parCapply, parLapply,
#> parLapplyLB, parRapply, parSapply, parSapplyLB
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, append, as.data.frame, basename, cbind,
#> colMeans, colnames, colSums, dirname, do.call, duplicated,
#> eval, evalq, Filter, Find, get, grep, grepl, intersect,
#> is.unsorted, lapply, lengths, Map, mapply, match, mget, order,
#> paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind,
#> Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
#> table, tapply, union, unique, unsplit, which, which.max,
#> which.min
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:base':
#>
#> expand.grid
#> Loading required package: IRanges
#>
#> Attaching package: 'IRanges'
#> The following object is masked from 'package:grDevices':
#>
#> windows
#> Loading required package: GenomicRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: SummarizedExperiment
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#> Loading required package: DelayedArray
#> Loading required package: matrixStats
#>
#> Attaching package: 'matrixStats'
#> The following objects are masked from 'package:Biobase':
#>
#> anyMissing, rowMedians
#> Loading required package: BiocParallel
#>
#> Attaching package: 'DelayedArray'
#> The following objects are masked from 'package:matrixStats':
#>
#> colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRanges
#> The following objects are masked from 'package:base':
#>
#> aperm, apply
dds <- DESeqDataSetFromMatrix(data, replicates, ~ replicates)
#> converting counts to integer mode
cts <- counts(dds)
geoMeans <- apply(cts, 1, function(row) if (all(row == 0)) 0 else exp(mean(log(row[row != 0]))))
dds <- estimateSizeFactors(dds, geoMeans=geoMeans)
#> Error in .local(object, ..., value): all(!is.na(value)) is not TRUE
norm <- counts(dds, normalized=TRUE)
#> Error in .local(object, ...): first calculate size factors, add normalizationFactors, or set normalized=FALSE
logcounts <- log2( counts(dds, normalized=TRUE) + 1 )# in case you want a log transformed normalized count
#> Error in .local(object, ...): first calculate size factors, add normalizationFactors, or set normalized=FALSE
write.csv(logcounts, file = "~/RSTUDIO/Bacteria-total/bacteria-total-normalizados.csv")
#> Error in is.data.frame(x): objeto 'logcounts' no encontrado
Created on 2020-02-06 by the reprex package (v0.3.0)
Inglés
Quisiste decir:
No puedo pegar mi base de datos en el formato que ustedes lo requieren REDEX, porque me arrojó este error R
I could not paste my database in the format that you require REDEX, because I threw this error R: R code execution error
But this is the image of my data base:
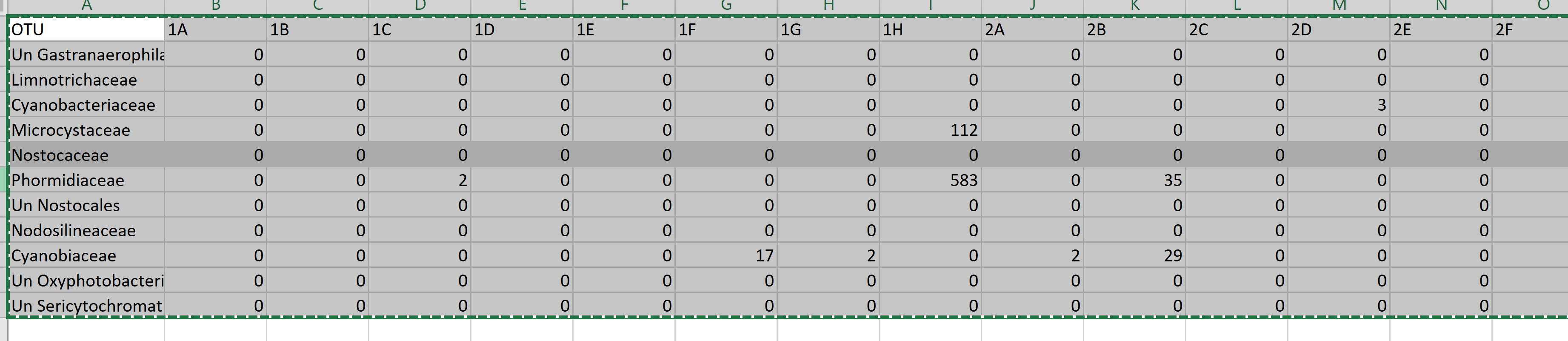
Thank you so much!