Metaboseek
Authors: Max Helf
Working with Shiny more than 1 year
Abstract: Many scientific R packages are of critical importance to their research communities, but don't have a graphical user interface. This makes them not only harder to access for beginners, and generally complicates browsing analysis results in a human-friendly way. To address this problem in the field of Metabolomics (the study of the small molecules in life), I wrote Metaboseek as a way to access the most popular computational workflows in our lab, most notably the xcms package. It first started as a simple R console script to inspect chromatographic peaks, but then grew into a shiny app with more and more functionality - this progression may sound familiar to many of you :)! Metaboseek now consists of about 70 shiny modules and there is extensive documentation for the app and the source code.
Full Description:
Some background
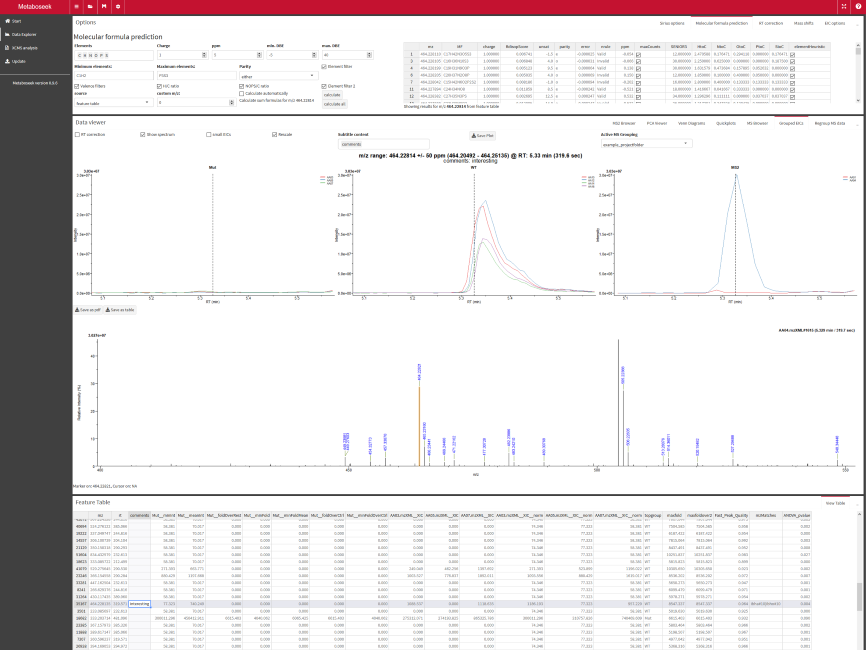
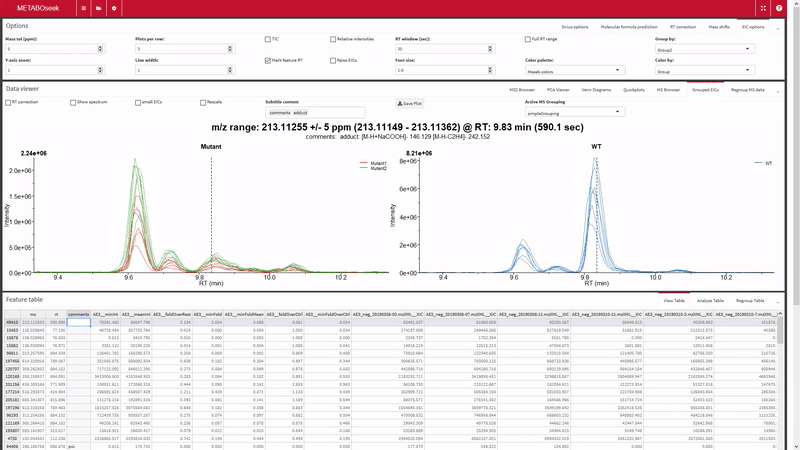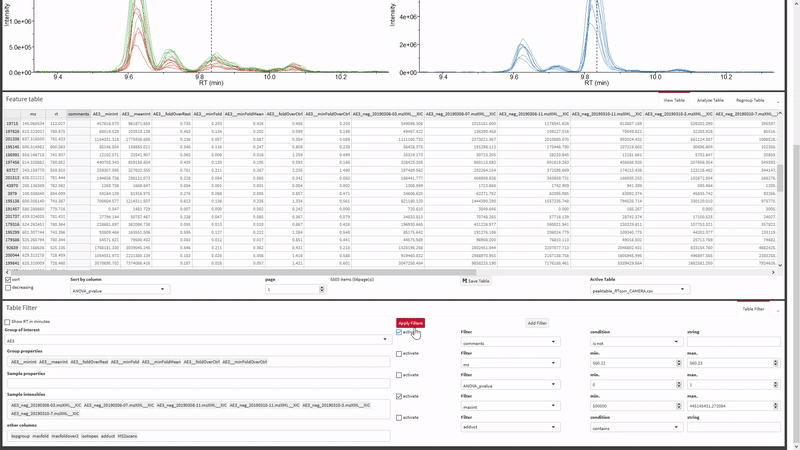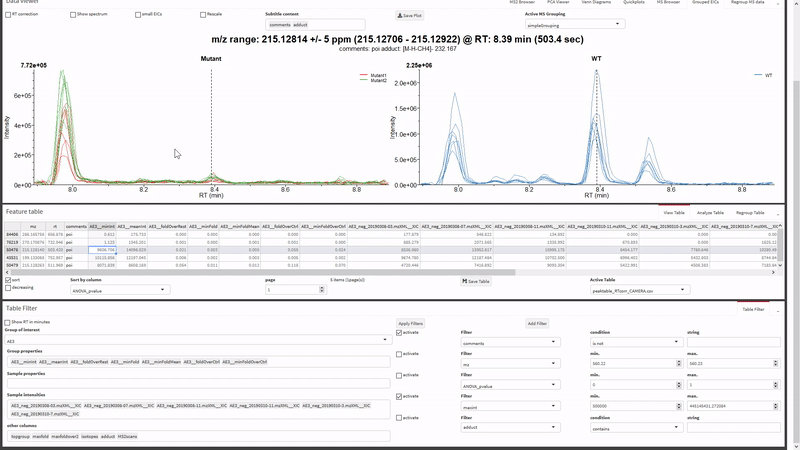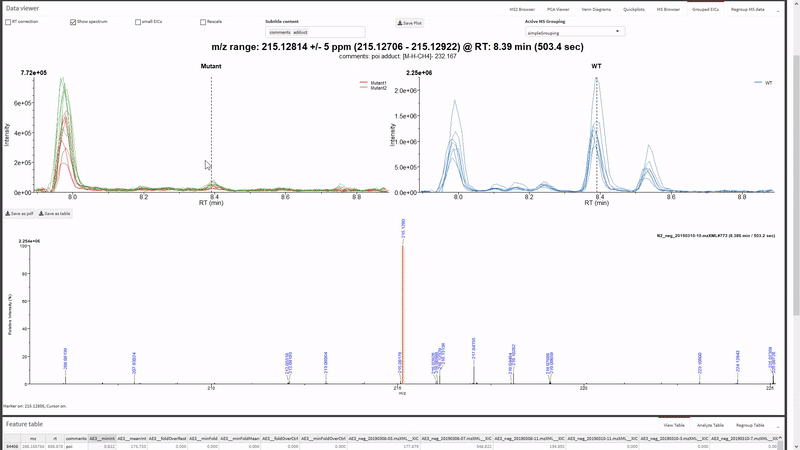
In Liquid Chromatography/Mass Spectrometry (LC/MS), so called mass spectra are acquired in rapid succession. Each spectrum can contain the mass-to-charge ratios (m/z) of thousands of charged molecules (ions) and their intensity (=relative abundance) at one time point. The molecules are separated through chromatography before entering the mass spectrometer, so that ions are separated in an additional dimension, the retention time (rt). This is particularly important to get separate signals for molecules with the same or very similar m/z value, but different structures (this happens a lot).
The combination of m/z value and retention time can be defined as a molecular feature. Although these molecular features typically don't allow unambigous identification of specific compounds, their abundance can be compared between different biological samples. This can be very useful, because a molecular feature might be up- or downregulated in one sample group compared to another. For instance, some molecular features might only be found in a mutant strain of bacteria, but not in the wild-type control. In the approach to metabolomics presented here, we can select such molecular features of interest and then try to get more information about the molecular structure by tandem-MS, a method that breaks molecules into pieces and then reports the m/z of the resulting fragments.
Features
So what can you do with this app? I would like to highlight some of the things it can do, and invite you to check out the example data that is loaded in the online version (and is also included in the Metaboseek R package). Documentation on how to use the app is available at Metaboseek.
Metaboseek offers a graphical user interface to set up data analysis with the xcms package to detect and align molecular features from LC/MS data across multiple samples. You can then load xcms results into the app as a "Feature Table" (using xcms and MSnbase packages, mzR-based) and run statistical analyses to identify molecular features of interest.
- Filter the xcms results, view and export chromatograms and mass spectra for molecular features of interest.
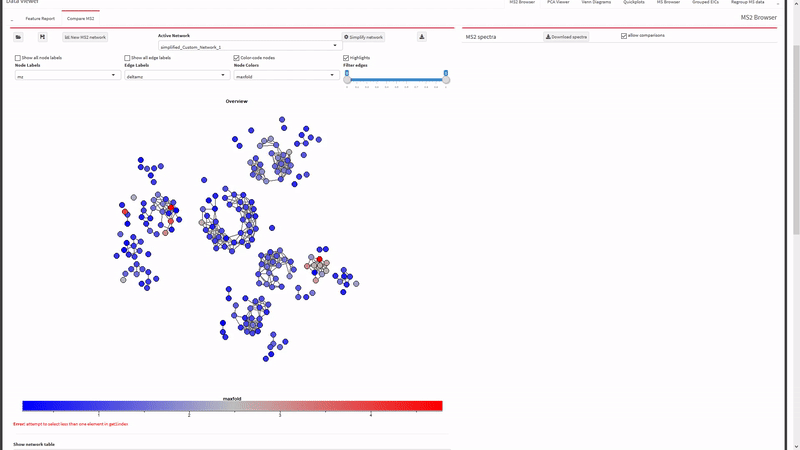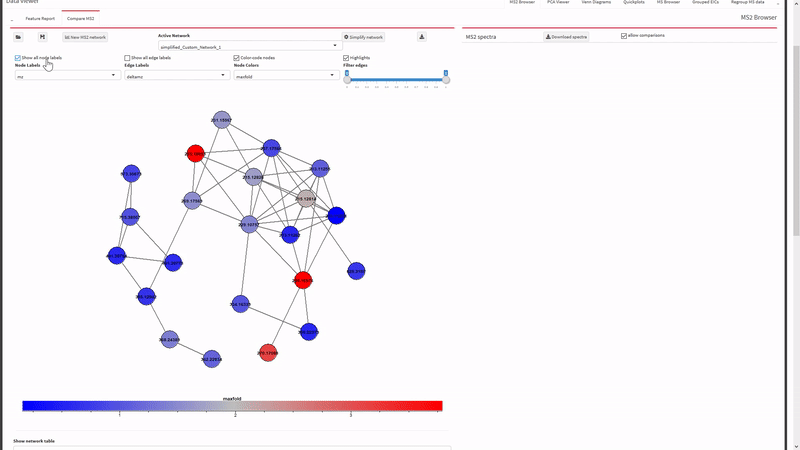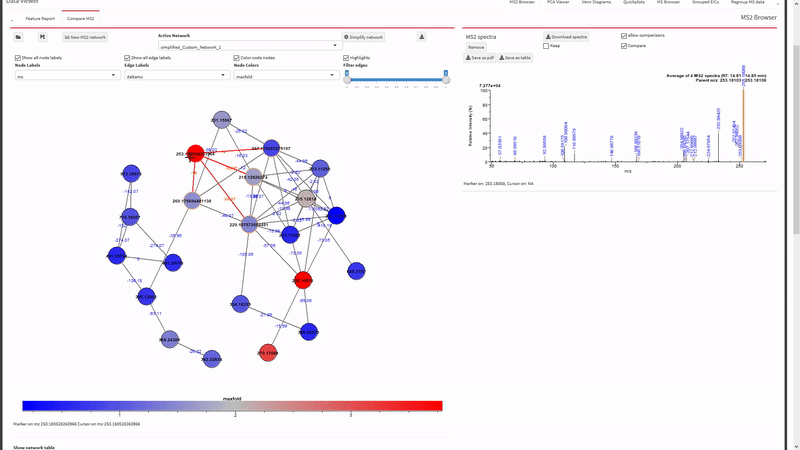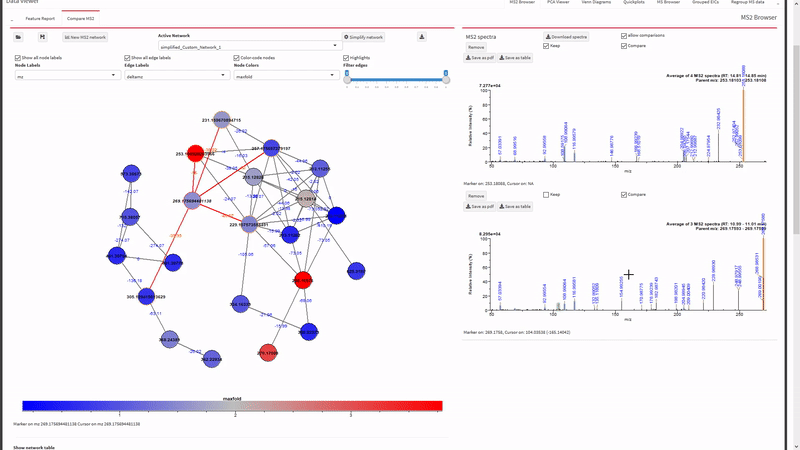
- Generate and view molecular networks based on tandem-MS spectrum similarity between molecular features (using
MassToolsandigraphpackages). The interactive plot is based on re-rendering plot.igraph() with different parameters depending on selections.
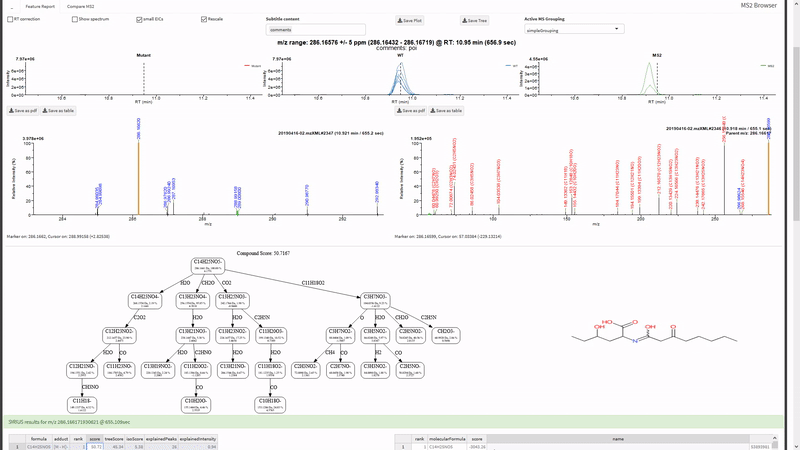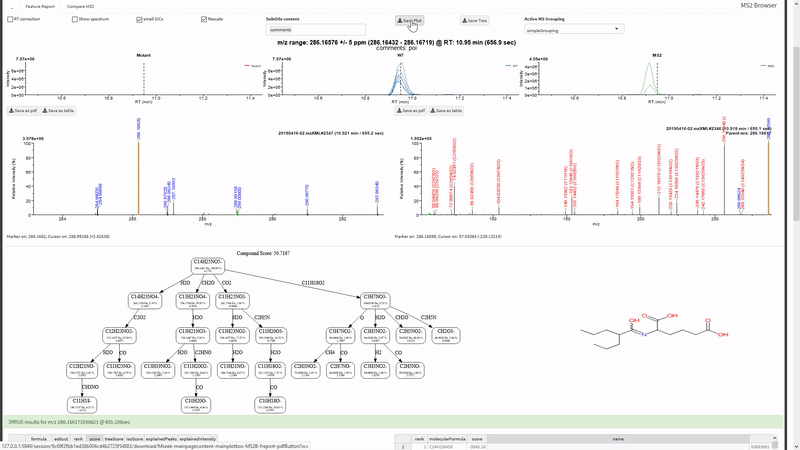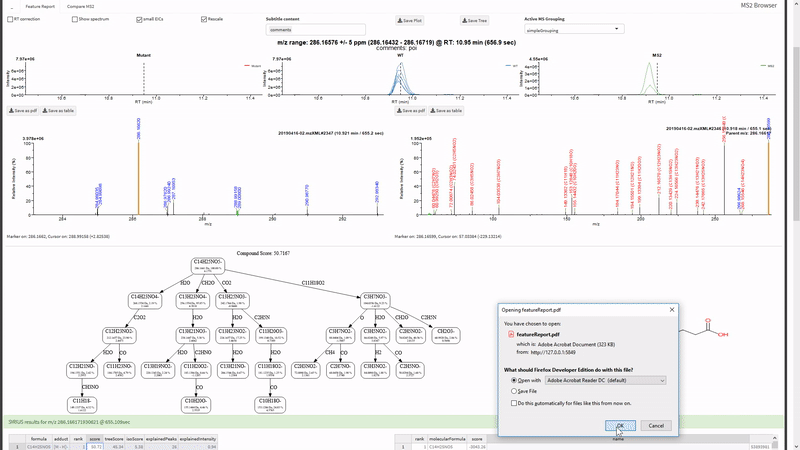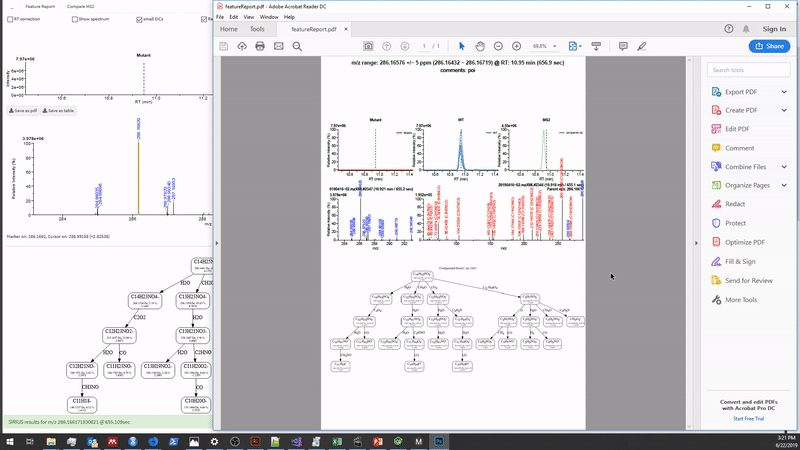
- Annotate fragments in tandem-MS spectra with SIRIUS. Metaboseek sends jobs to the SIRIUS command line interface and retrieves them from the SIRIUS output folder.
Shiny Details
Here are some of the a technical perspective, I would like to Metaboseek is strictly built from shiny modules, allowing for easy reuse and rearrangement of elements such as the spectral plots
-
Metaboseek is an R package that contains the app code along with some data analysis functions
-
For improved code organization, Metaboseek is built from more than 70 different shiny modules, all of which are part of the Metaboseek package and documented. I am considering splitting grouping these modules into sub-packages for easier reuse by others.
-
a
reactivevaluesobject calledvaluesis generated in the app, and is passed as an argument into the larger modules. Because elements inreactivevaluesare mutable, changing them in one module has side effects that can be observed across the app. Some commonly accessed values are safely accessed by getter and setter functions (e.g. making sure the latest edits to theFeature Tableare applied before getting its contents). This makes it easy to rearrange module hierarchy and makes the central data editable from all parts of the app. I would be happy to discuss this more and have some thoughts, e.g. here. -
You can use your arrow keys to browse through the
Feature Tableand switch between plots (for spectra and chromatograms) very quickly. To make this run smoothly, I had to make some compromises. For plotting, I use base-R plots.ggplot(andplotly) take longer to build the plot when the plotting data is changed, and browsing the Feature Table would be slow. In addition, the plotted data is drawn from the LC/MS data that is loaded into the session, and currently that means it is in memory and accessed through (deprecated)xcmsfunctions which work faster than the current functions in this use case (getting a small amount of data from many files at a time). -
The central object of class
MseekFTcontains theFeature Tablealong with its processing history. It can be saved from the UI (usingsaveRDS()internally) so that upon reloading the data, all previous steps are on record. The processing history can also be viewed in the app. AllMseekFTobjects in a session can be saved ("Save session") to facilitate sharing of information or resuming work. -
Data is generally loaded from and saved to the server side (and accessed with the
shinyFilespackage). This avoids overhead from uploading the very large MS data files or, when running the app locally on a workstation, unnecessarily copying them.
Deployment
Unfortunately, I was not able to deploy the app on shinyapps.io. There seems to be a problem with installing one of the required packages (mzR). However, there are other options to try the app:
-
Here is the direct link to the app (with example data pre-loaded), hosted on a shiny-server instance at Boyce Thompson Institute/Cornell University (on this server, it takes up to a minute to load; when installed locally, it starts within a few seconds)
-
Take a look at the documentation here to see how to get Metaboseek as installer or .zip file for Windows (built using R-portable), as a Docker image or from GitHub.
I would be excited to get some feedback and suggestions on the app, and I will be happy to answer your questions!
Acknowledgements
Thanks to the Boyce Thompson Institute/Cornell University for their support, and to Prof. Frank Schroeder, Dr. Bennett Fox and the entire Schroeder lab for testing the app and suggesting features.
Edit: Please note the improved documentation at Metaboseek
Category: Research
Keywords: data analysis, research, mass spectrometry, metabolomics, BYOD, shiny modules
Shiny app: http://mosaic.bti.cornell.edu/Metaboseek/
Repo: GitHub - mjhelf/Metaboseek: Interactive software to analyze and browse mass spectrometry data
RStudio Cloud: Posit Cloud
Thumbnail:

Full image: