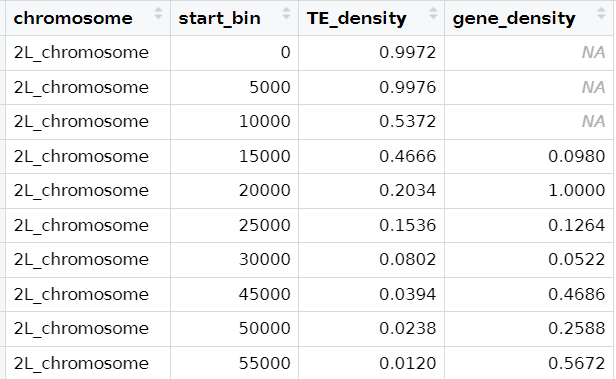
I want to bind two tables based on column 1 and 2. however there are rows that are missing. I wan all bins to be included in the merged table.
I tried:
combined <- full_join(TE_eu, gene_eu, by = c("chromosome", "start_bin"))
and noticed that it only includes 'start_bin' column from the first data frame, and the bins that exist in table 2 are missing e.g. 30000, 40000
I tried using bind_rows, but it doesn't quite fit with my purpose as it basically just adds the two table in one file and does not merge the rows.
Can you post the output that you get for combined with the current line of code?
Because for me this line works perfectly on these 2 tables (assuming they are regular dataframes?).
However, instead of zeros, you will get NAs where there are no data for that row/column, so you will have to replace them with zeros. Be careful with that though, as there is a different meaning behind those (NA = not measured/do not have that info, 0 = it was measured and the actual result was 0).
I am trying to use your code, in which part of the library is replace_na?
Joining with by = join_by(chromosome, start_bin)
Error in mutate(): In argument: across(ends_with("density"), function(x) replace_na(x, 0)).
Caused by error in across():
! Can't compute column TE_density.
Caused by error in replace_na():
! could not find function "replace_na"
Run rlang::last_trace() to see where the error occurred.