Hi,
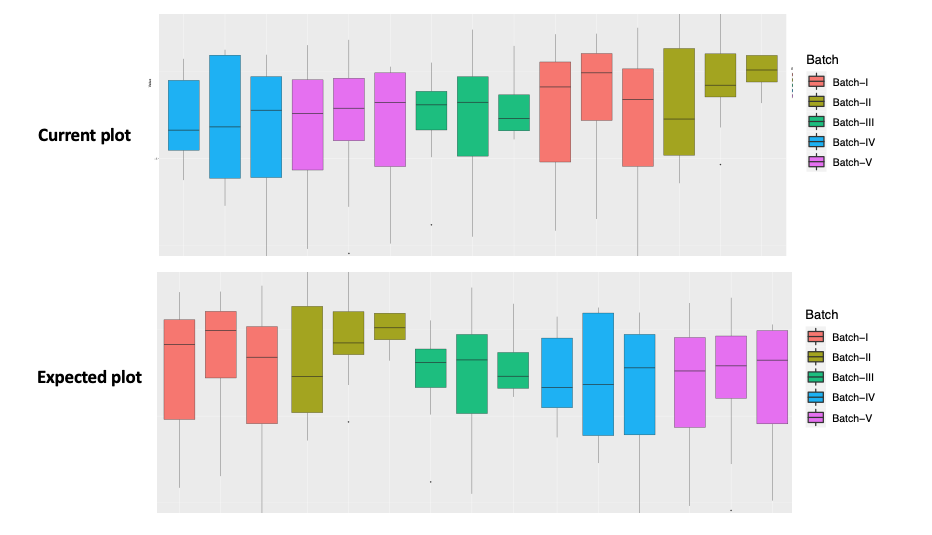
I have a question to arrange the x-axis in the Boxplot through ggplot2 (geom_boxplot), but not successful. Actually trying to arrange as (Batch I, Batch II, ........Batch V). Please provide inputs how to fix this issue.
dput(Data_v1)
structure(list(ID = c("CH30_0h-1", "CH30_6h-8", "CH30_6h-16",
"CN1_0h-1", "CN1_6h-8", "CN1_6h-16", "400B_0h-1", "400B_6h-8",
"400B_6h-16", "1111A_0h-1", "1111A_6h-8", "1111A_6h-16", "1164A_0h-1",
"1164A_6h-8", "1164A_6h-16"), Gene_1 = c(-7.06771, -8.73071,
-6.73071, -4.73797, NA, NA, -6.89689, -9.11833, -7.24038, -9.91257,
-7.31379, -6.17557, -7.58907, -7.43676, NA), Gene_2 = c(-5.91461,
-6.36411, -5.02691, -9.64877, NA, -4.1052, -4.9579, -4.44811,
-4.94366, -5.61523, -5.6951, -5.6236, -5.61506, -5.724, -6.38191
), Gene_3 = c(-2.1792, -2.3658, -2.02961, -1.26177, -2.03669,
-2.4839, -2.24417, -1.76436, -1.29733, -2.30767, -2.57192, -1.87333,
-2.72559, -2.36064, -1.59041), Gene_4 = c(-4.24441, -3.40551,
-1.96791, -4.46717, -2.54759, -0.8027, -4.17766, -3.21896, -2.26203,
-4.00953, -3.06621, -2.15282, -4.45781, -3.77418, -4.48653),
Gene_5 = c(-5.38011, -5.66761, -3.26111, -5.70527, 4.26221,
-5.1683, NA, NA, NA, -4.78791, -8.95204, -6.35331, -6.34912,
-5.70766, -7.71973), Gene_6 = c(-2.36521, -2.59891, -2.15781,
-0.75477, -2.03779, -1.196, -3.06798, -3.85273, -2.89402,
-2.13551, -2.60488, -1.93915, -2.68515, -3.7548, -2.69344
), Gene_7 = c(-2.05641, -3.8735, -2.91651, -2.66677, -3.40529,
-2.8928, -3.4558, -3.89929, -3.38236, -3.92386, -4.15504,
-3.77592, -3.96861, -2.36629, -3.30789), Gene_8 = c(-1.4283,
-1.79241, -1.75011, -3.25977, -2.36509, -1.9922, -2.53121,
-2.58246, -2.649, -2.36292, -2.0177, -1.99138, -1.74274,
-3.02425, -2.16307), Gene_9 = c(-3.32931, -3.20471, -4.11671,
-4.95547, -2.76539, -3.227, -3.90394, -4.20117, -4.92701,
-4.67031, -5.08027, -5.37573, -3.43241, -2.51116, -3.55333
), Gene_10 = c(-2.55501, -1.24421, -1.41321, -1.56437, -2.90679,
-3.2299, -3.31768, -3.16836, -4.02685, -4.35349, -2.77335,
-4.40128, -2.77479, -2.61881, -3.58733)), class = "data.frame", row.names = c("CH30_0h-1",
"CH30_6h-8", "CH30_6h-16", "CN1_0h-1", "CN1_6h-8", "CN1_6h-16",
"400B_0h-1", "400B_6h-8", "400B_6h-16", "1111A_0h-1", "1111A_6h-8",
"1111A_6h-16", "1164A_0h-1", "1164A_6h-8", "1164A_6h-16"))
#> ID Gene_1 Gene_2 Gene_3 Gene_4 Gene_5 Gene_6
#> CH30_0h-1 CH30_0h-1 -7.06771 -5.91461 -2.17920 -4.24441 -5.38011 -2.36521
#> CH30_6h-8 CH30_6h-8 -8.73071 -6.36411 -2.36580 -3.40551 -5.66761 -2.59891
#> CH30_6h-16 CH30_6h-16 -6.73071 -5.02691 -2.02961 -1.96791 -3.26111 -2.15781
#> CN1_0h-1 CN1_0h-1 -4.73797 -9.64877 -1.26177 -4.46717 -5.70527 -0.75477
#> CN1_6h-8 CN1_6h-8 NA NA -2.03669 -2.54759 4.26221 -2.03779
#> CN1_6h-16 CN1_6h-16 NA -4.10520 -2.48390 -0.80270 -5.16830 -1.19600
#> 400B_0h-1 400B_0h-1 -6.89689 -4.95790 -2.24417 -4.17766 NA -3.06798
#> 400B_6h-8 400B_6h-8 -9.11833 -4.44811 -1.76436 -3.21896 NA -3.85273
#> 400B_6h-16 400B_6h-16 -7.24038 -4.94366 -1.29733 -2.26203 NA -2.89402
#> 1111A_0h-1 1111A_0h-1 -9.91257 -5.61523 -2.30767 -4.00953 -4.78791 -2.13551
#> 1111A_6h-8 1111A_6h-8 -7.31379 -5.69510 -2.57192 -3.06621 -8.95204 -2.60488
#> 1111A_6h-16 1111A_6h-16 -6.17557 -5.62360 -1.87333 -2.15282 -6.35331 -1.93915
#> 1164A_0h-1 1164A_0h-1 -7.58907 -5.61506 -2.72559 -4.45781 -6.34912 -2.68515
#> 1164A_6h-8 1164A_6h-8 -7.43676 -5.72400 -2.36064 -3.77418 -5.70766 -3.75480
#> 1164A_6h-16 1164A_6h-16 NA -6.38191 -1.59041 -4.48653 -7.71973 -2.69344
#> Gene_7 Gene_8 Gene_9 Gene_10
#> CH30_0h-1 -2.05641 -1.42830 -3.32931 -2.55501
#> CH30_6h-8 -3.87350 -1.79241 -3.20471 -1.24421
#> CH30_6h-16 -2.91651 -1.75011 -4.11671 -1.41321
#> CN1_0h-1 -2.66677 -3.25977 -4.95547 -1.56437
#> CN1_6h-8 -3.40529 -2.36509 -2.76539 -2.90679
#> CN1_6h-16 -2.89280 -1.99220 -3.22700 -3.22990
#> 400B_0h-1 -3.45580 -2.53121 -3.90394 -3.31768
#> 400B_6h-8 -3.89929 -2.58246 -4.20117 -3.16836
#> 400B_6h-16 -3.38236 -2.64900 -4.92701 -4.02685
#> 1111A_0h-1 -3.92386 -2.36292 -4.67031 -4.35349
#> 1111A_6h-8 -4.15504 -2.01770 -5.08027 -2.77335
#> 1111A_6h-16 -3.77592 -1.99138 -5.37573 -4.40128
#> 1164A_0h-1 -3.96861 -1.74274 -3.43241 -2.77479
#> 1164A_6h-8 -2.36629 -3.02425 -2.51116 -2.61881
#> 1164A_6h-16 -3.30789 -2.16307 -3.55333 -3.58733
dput(Sample_Pheno)
structure(list(ID = c("CH30_0h-1", "CH30_6h-8", "CH30_6h-16",
"CN1_0h-1", "CN1_6h-8", "CN1_6h-16", "400B_0h-1", "400B_6h-8",
"400B_6h-16", "1111A_0h-1", "1111A_6h-8", "1111A_6h-16", "1164A_0h-1",
"1164A_6h-8", "1164A_6h-16"), Batch = c("Batch-I", "Batch-I",
"Batch-I", "Batch-II", "Batch-II", "Batch-II", "Batch-III", "Batch-III",
"Batch-III", "Batch-IV", "Batch-IV", "Batch-IV", "Batch-V", "Batch-V",
"Batch-V")), class = "data.frame", row.names = c(NA, -15L))
#> ID Batch
#> 1 CH30_0h-1 Batch-I
#> 2 CH30_6h-8 Batch-I
#> 3 CH30_6h-16 Batch-I
#> 4 CN1_0h-1 Batch-II
#> 5 CN1_6h-8 Batch-II
#> 6 CN1_6h-16 Batch-II
#> 7 400B_0h-1 Batch-III
#> 8 400B_6h-8 Batch-III
#> 9 400B_6h-16 Batch-III
#> 10 1111A_0h-1 Batch-IV
#> 11 1111A_6h-8 Batch-IV
#> 12 1111A_6h-16 Batch-IV
#> 13 1164A_0h-1 Batch-V
#> 14 1164A_6h-8 Batch-V
#> 15 1164A_6h-16 Batch-V
library(reshape2)
Data_v1_melt <- melt(Data_v1,
id.vars = "ID",
variable.name = "Genes",
value.name = "Value")
Data_v1_merged <- merge(Data_v1_melt, Sample_Pheno, by = "ID", all = TRUE)
head(Data_v1_merged)
Data_v1_merged = Data_v1_merged[order(Data_v1_merged$Batch),]
str(Data_v1_merged)
Plot_type <- "test"
library(ggplot2)
pdf(paste0(Plot_type,"_Testing_onlyxxx.pdf"),height = 25,width = 30)
p <- ggplot(Data_v1_merged) +
geom_boxplot(aes(x=ID, y=Value, fill=Batch))
plot(p)
dev.off()
Created on 2022-01-06 by the reprex package (v2.0.1)
