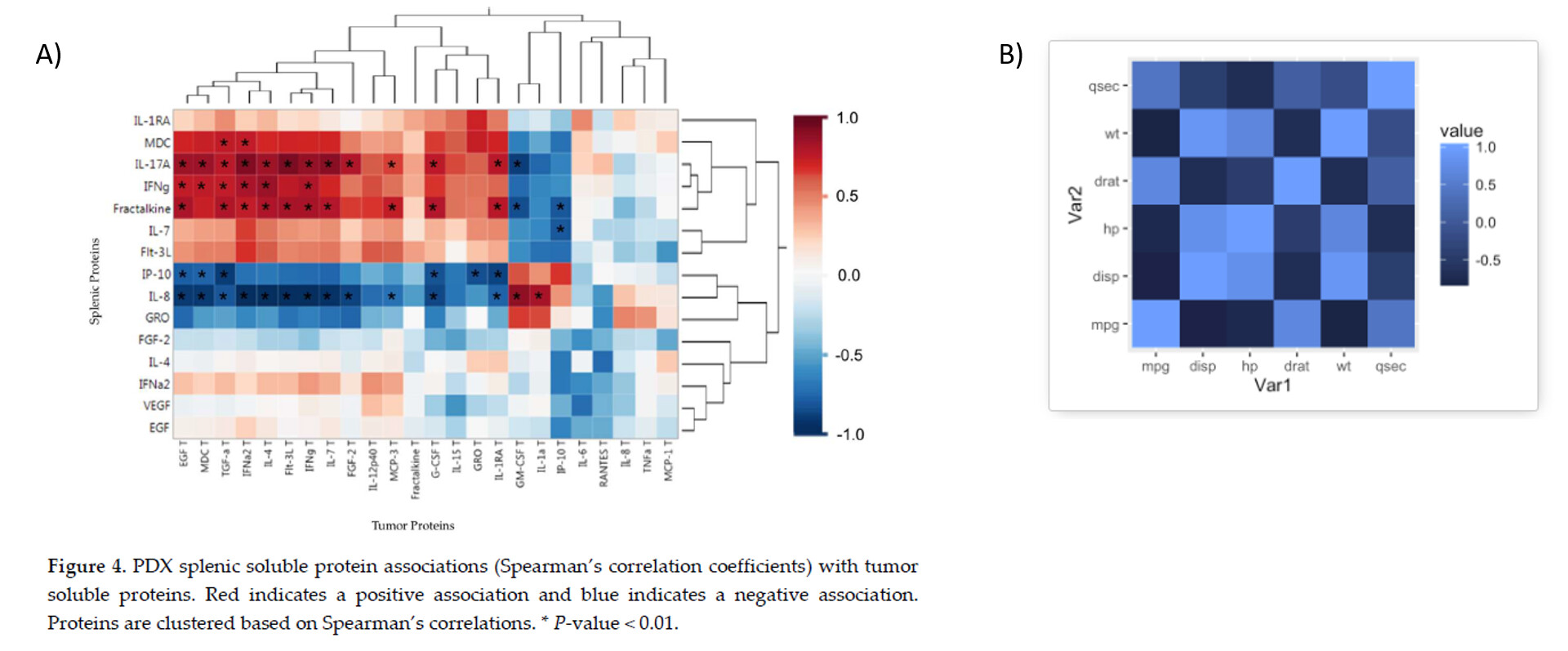
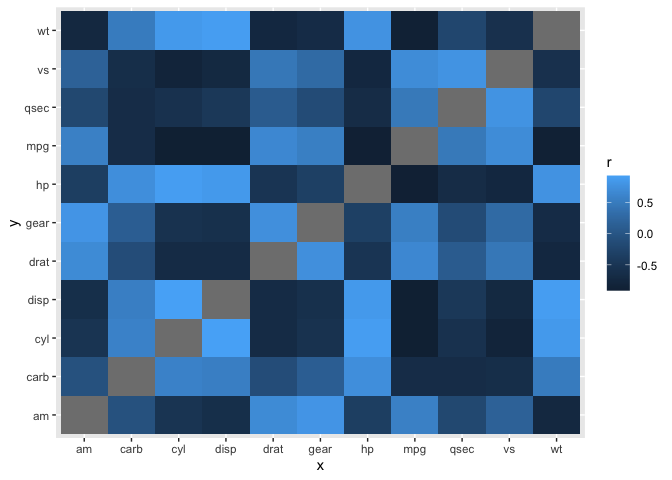
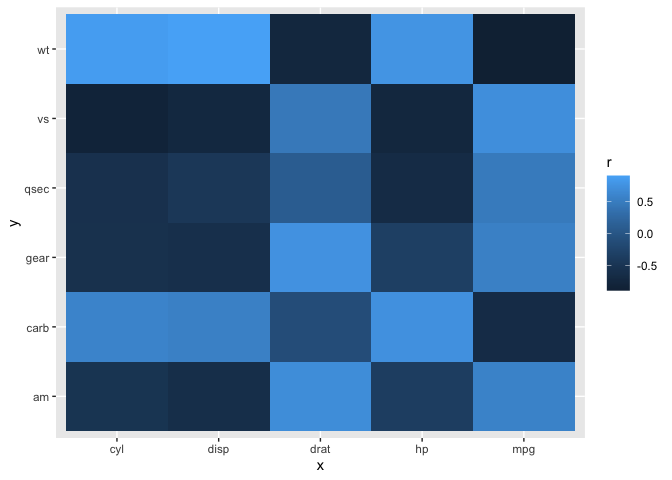
It's basically a matter of filtering your x and y axes to suite your needs after computing all pair-wise correlations using standard methods (I used corrr here). Here is a simplified example of how the heatmap would look without filtering (similar to Figure B) and after filtering the x and y axis to show different variables on each axis (similar to Figure A). You could rework this code to have all of the gene expression variables on one axis and protein expression on the other.
library(corrr)
library(dplyr)
library(ggplot2)
sp <-
mtcars %>%
correlate(method = "spearman") %>%
stretch()
# Like Figure B
sp %>%
ggplot(aes(x, y, fill = r)) +
geom_tile()
# Filter based on what variables you want on the
# x and y axes for the heatmaps
sp_filtered <-
sp %>%
filter(
x %in% c("mpg", "cyl", "disp", "hp", "drat"),
y %in% c("wt", "qsec", "vs", "am", "gear", "carb")
)
# Like Figure A
sp_filtered %>%
ggplot(aes(x, y, fill = r)) +
geom_tile()