Regression models in the gamlss package have an associated distribution family such as NO or BCCG or BCT. Here is a BCCG model of height vs age and the fitted centiles:
suppressPackageStartupMessages({
library(dplyr)
library(tibble)
library(gamlss)
library(glue)
library(AGD)
})
data <-AGD::boys7482[, c('age', 'hgt')]
data <- na.omit(data[sample(nrow(data), 500), ])
object <- gamlss(hgt ~ pbm(age), sigma.form=~pb(age), family=BCCG,
data=data, trace=FALSE)
centiles(object, data$age, xlab='age', ylab='height')
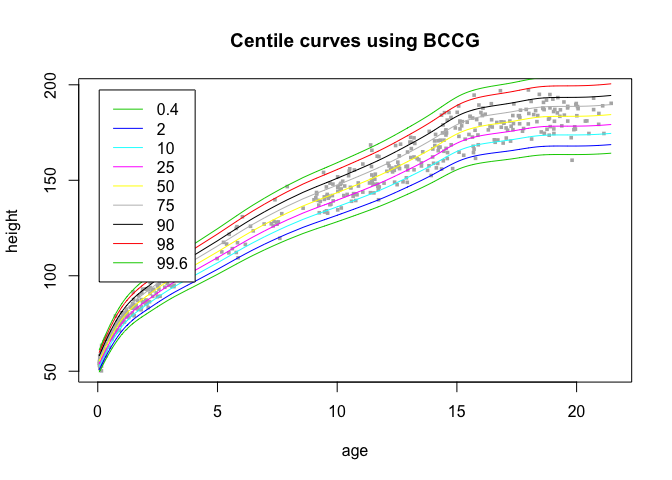
#> % of cases below 0.4 centile is 0.4081633
#> % of cases below 2 centile is 1.836735
#> % of cases below 10 centile is 10.61224
#> % of cases below 25 centile is 24.69388
#> % of cases below 50 centile is 47.55102
#> % of cases below 75 centile is 75.5102
#> % of cases below 90 centile is 89.59184
#> % of cases below 98 centile is 97.7551
#> % of cases below 99.6 centile is 99.79592
The parameters for each model family are a subset of c('mu', 'sigma', 'nu', 'tau') and are extracted as follows:
newage <- tibble(age = 0:20)
(pars <- as_tibble(predictAll(object, newage, data=data)))
#> new prediction
#> New way of prediction in pbm() (starting from GAMLSS version 5.0-3)
#> new prediction
#> New way of prediction in pb() (starting from GAMLSS version 5.0-3)
#> # A tibble: 21 x 3
#> mu sigma nu
#> <dbl> <dbl> <dbl>
#> 1 53.5 0.0394 0.719
#> 2 77.1 0.0395 0.719
#> 3 88.9 0.0397 0.719
#> 4 97.8 0.0398 0.719
#> 5 105. 0.0399 0.719
#> 6 112. 0.0400 0.719
#> 7 120. 0.0401 0.719
#> 8 127. 0.0403 0.719
#> 9 133. 0.0404 0.719
#> 10 139. 0.0405 0.719
#> # … with 11 more rows
For BCCG the parameters are c('mu', 'sigma', 'nu'), i.e. names(pars).
There are then functions to process the parameters, with the general name [dpqr]family such as:
# qNO(p, mu, sigma)
# pBCCG(q, mu, sigma, nu)
# rBCT(n, mu, sigma, nu, tau)
but they all have different numbers of arguments. I'd like to write code to handle these cases generally in a dplyr thread. I can extract e.g. the qfamily function as follows, and this works for all families:
qfamily <- eval(parse(text = glue('gamlss.dist::q{object$family[[1]]}')))
Then I can process the data like this, so long as I explicitly name the parameters:
bind_cols(newage, pars) %>%
mutate(p10 = qfamily(0.1, mu=mu, sigma=sigma, nu=nu))
#> # A tibble: 21 x 5
#> age mu sigma nu p10
#> <int> <dbl> <dbl> <dbl> <dbl>
#> 1 0 53.5 0.0394 0.719 50.8
#> 2 1 77.1 0.0395 0.719 73.2
#> 3 2 88.9 0.0397 0.719 84.4
#> 4 3 97.8 0.0398 0.719 92.8
#> 5 4 105. 0.0399 0.719 99.8
#> 6 5 112. 0.0400 0.719 107.
#> 7 6 120. 0.0401 0.719 114.
#> 8 7 127. 0.0403 0.719 121.
#> 9 8 133. 0.0404 0.719 126.
#> 10 9 139. 0.0405 0.719 132.
#> # … with 11 more rows
This works for a BCCG object but not for NO or BCT.
So what I'm looking for is a mutate incantation, possibly including vars(names(pars)), that will replace the mu=mu, sigma=sigma, nu=nu list in the call and generalise it to all families.
Created on 2019-11-22 by the reprex package (v0.3.0)