Hello,
I would like to use Rstudio to retrieve specific sequences by their known positions from a available genome sequence.
I have the complete genome sequence downloaded available in a fasta format, which contains the sequences of all 12 chromosomes. Below you see a representation of this file:
>ST4.03ch01
CCATTTTAAAGGTCAAATGTGACCCAGAGCAGGCAAAACCCAAATTTTATCGATTTTCGTGTGCAATAGT
CTATGGAGTTTTTGGTGATCTGGAATTCCGACATAAGTTATGCTAAAAAATTTTGTGTACGTCTGTTAAG
ACCTTAGCTATGAAGCCAGTTAGCCCTCACGGCCAAAACATACCATTTTGAAGGTCAAATATGCCCCGAA
TCTGGTAAACCCCCCAATTTGCCGATTATCATGTGCTATAGTCCATGGACTTTTTGGTGATCTGGAATTT
CGACATACTTTTTGCCAAAAATTTTCGTACACTTCCGTTAAAACCTTAGCTATGGATCCAGTTAGACTTC
(etc...)
>ST4.03ch02
ACCAACAATTTACCATAATTAAGGCATAGATAATGATGCAATTCAATAACCGCAAAGCAATTTAGACATA
TTCAAATCACAAGCTTCCATAATCAGCAAACCCACTTCATAAAGACACAATTTAGGAATTGAAAGAAATC
ATGGGTTCATAGAGTATTTTCATCATAAATCATTAATTAAACACTAATACAATCATAGTATGACTTTAGT
AACCATTTGAAATCATTTGGAGAAAGAACCCATGAGATTGAGTAAGACCCTAGGTTTTTCATGAACTTGA
AAACTTTGAAAACTTCCTTGAAATTGACTTTAGGGGTGAAAGCTACCCCTAGATGAAGGATCACCATACC
TTTGCTAAGATTTCCCAAGAAATTTGATGAAGAAACGCCTTGAGCTTCAATGGATCCTTTCTTCTTCTTC
(etc...)
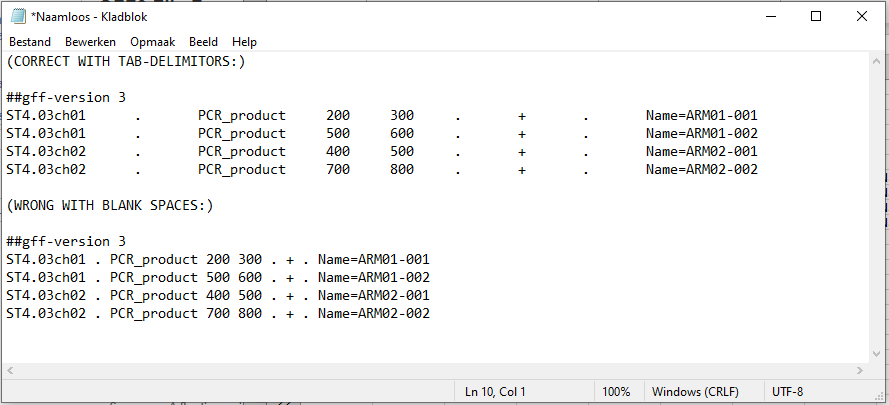
I have a tab-delimited GFF3 file with the start and end coordinates of all the sequences I would like to retrieve , see the top of this file below.
|ST4.03ch01|.|PCR_product|5105452|5105605|.|+|.|Name=ARM01-004;Note=validated marker|
|ST4.03ch01|.|PCR_product|5319044|5319174|.|+|.|Name=ARM01-005;Note=validated marker|
|ST4.03ch01|.|PCR_product|5319162|5319313|.|+|.|Name=ARM01-006;Note=validated marker|
|ST4.03ch01|.|PCR_product|5319288|5319529|.|+|.|Name=ARM01-007;Note=validated marker|
|ST4.03ch01|.|PCR_product|5319526|5319779|.|+|.|Name=ARM01-008;Note=validated marker|
Column 1 corresponds to the chromosome numbers of the fasta genome file.
Column 4&5 correspond to the start and end position of the desired sequence.
Column 9 is filled with a name and a note, separated by semicolons according to GFF3 specifications.
(empty columns are filled with "." according to GFF3 specifications.)
I know there are several bio-packages like ape or biostrings, but I have not figured out yet how to make this work.
My desired output would look like this (name and sequence):
>ARM01-004
CCATTTTAAAGGTCAAATGTGACCCAGAGCAGGCAAAACCCAAATTTTATCGATTTTCGTGTGCAATAGT
CTATGGAGTTTTTGGTGATCTGGAATTCCGACATAAGTTATGCTAAAAAATTTTGTGTACGTCTGTTAAG
ACCTTAGCTATGAAGCCAGTTAGCCCTCACGGCCAAAACATACCA
>ARM01-005
TCTGGTAAACCCCCCAATTTGCCGATTATCATGTGCTATAGTCCATGGACTTTTTGGTGATCTGGAATTT
CGACATACTTTTTGCCAAAAATTTTCGTACA
>ARM01-006
CCATTTTAAAGGTCAAATGTGACCCAGAGCAGGCAAAACCCAAATTTTATCGATTTTCGTGTGCAATAGT
CTATGGAGTTTTTGGTGATCTGGAATTCCGACATAAGTTATGCTAAAAAATTTTGTG
etc...
Note that the output sequences shown above are dummies, as for example.
Thanks for any suggestions.