Hi, I have trouble with dot_sankey plot and need help with my script.
this is my data:
structure(list(Pathway = c("Alpha-amino acid biosynthetic proc. ",
"Carboxylic acid biosynthetic proc. ", "Organic acid biosynthetic proc. ",
"Small molecule biosynthetic proc. ", "Carboxylic acid metabolic proc. ",
"Oxoacid metabolic proc. ", "Organic acid metabolic proc. ",
"Small molecule metabolic proc. ", "Cellular catabolic proc. ",
"Catabolic proc. "), GeneRatio = c(0.133333333333333, 0.0668693009118541,
0.0664652567975831, 0.0558375634517767, 0.0427263479145473, 0.0426164519326065,
0.0418287937743191, 0.0377066115702479, 0.0324729392173189, 0.0299465240641711
), FDR = c(3.8517556300416, 4.94174664623985, 4.94174664623985,
5.91926984684473, 5.20765687373531, 5.20765687373531, 5.19008290917224,
7.78847331776209, 5.91926984684473, 5.20765687373531), GeneID = c("ASNS/CTH/ASS1/PSAT1/CBS/ATP2B4/OAT/PHGDH/MTHFD1/PLOD3",
"ASNS/CTH/ASS1/ACLY/PSAT1/OXSM/CBS/NA/CASP1/ATP2B4/MTHFD1/KYNU/OLAH/AKR1C3/OAT/PHGDH/PLOD3/HSD17B12/FABP5/ERLIN2/PRMT3/CBR1",
"ASNS/CTH/ASS1/ACLY/PSAT1/OXSM/CBS/NA/CASP1/ATP2B4/MTHFD1/KYNU/OLAH/AKR1C3/OAT/PHGDH/PLOD3/HSD17B12/FABP5/ERLIN2/PRMT3/CBR1",
"ASNS/COQ9/COQ5/CTH/COQ6/ASS1/ACLY/PSAT1/OXSM/CBS/FDXR/PC/NA/CASP1/ATP2B4/MTHFD1/KYNU/ERLIN2/OLAH/AKR1C3/OAT/PHGDH/PLOD3/ALDOC/HSD17B12/ADK/FABP5/PTPN2/PRMT3/SIRT5/AACS/ACSS3/CBR1",
"ACAA1/OAT/ASNS/ARG2/QPRT/FAH/ALDOC/KYNU/CTH/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/PC/AKR1C3/NA/DLD/CASP1/ATP2B4/MTHFD1/ALDH1L2/OLAH/FOXK1/ACAD9/GPX1/LYPLA2/AACS/PHGDH/PLOD3/SLC27A1/HSD17B12/FABP5/GFPT1/NUDT19/ERLIN2/PRMT3/GSS/CBR1",
"ACAA1/OAT/ASNS/ARG2/QPRT/FAH/ALDOC/KYNU/CTH/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/PC/AKR1C3/NA/DLD/CASP1/ATP2B4/MTHFD1/ALDH1L2/OLAH/FOXK1/ACAD9/GPX1/LYPLA2/AACS/PHGDH/PLOD3/SLC27A1/HSD17B12/FABP5/GFPT1/NUDT19/ERLIN2/PRMT3/GSS/ABHD14B/CBR1",
"ACAA1/OAT/ASNS/ARG2/QPRT/FAH/ALDOC/KYNU/CTH/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/PC/AKR1C3/NA/DLD/CASP1/ATP2B4/MTHFD1/ALDH1L2/OLAH/FOXK1/ACAD9/GPX1/LYPLA2/AACS/PHGDH/PLOD3/SLC27A1/HSD17B12/FABP5/GFPT1/NUDT19/ERLIN2/PRMT3/GSS/ABHD14B/CBR1",
"ACAA1/OAT/ASNS/ARG2/COQ9/MTHFD1/QPRT/FAH/NAMPT/ALDOC/COQ5/KYNU/CTH/COQ6/NUDT10/PPCS/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/FLAD1/FDXR/DCXR/PC/IMPDH2/FUCA1/CA13/AKR1C3/NUDT11/GFPT1/NA/DLD/SNX17/CASP1/AIFM2/ATP2B4/DGUOK/ALDH1L2/UCK2/ERLIN2/OLAH/CBR1/FOXK1/ACAD9/GPX1/BAD/LYPLA2/MAT2B/SCARB1/AACS/PHGDH/PLOD3/HDLBP/CAT/SLC27A1/ HSD17B12/ADK/FABP5/PTPN2/NUDT19/BRAT1/PRMT3/SIRT5/DIP2A/ERH/GSS/ACSS3/ABHD14B/NEIL2",
"ACAA1/OAT/NEDD4/ARG2/DIS3/ACHE/HMOX1/PYGL/TIMP1/QPRT/FAH/UBE2S/SKP1/KYNU/CLU/CAT/NUDT10/PSME3/HSD17B4/STAM/PCYOX1L/CLGN/FBXL18/AGL/ERAP1/FBXO22/TMUB2/GUSB/UBE2L3/LONP1/NUDT11/PSMB10/FIS1/GPX1/NA/HGS/LYPLA2/ATP2B4/GPC1/FAP/DLD/PON2/WFS1/RBM24/OPTN/RBM38/HSPBP1/GSTM3/ALDH1L2/TGFB1I1/ERLIN2/DFFA/CBS/FOXK1/BCL2/TBK1/AKR1C3/SCARB1/SRPX/UCHL3/IDH1/RCN3/EPHX1/NUDT19/STX12/UFM1/ITGB4/CPTP/METTL3/FUCA1/SNX5/BAD/HAX1/PRKCA/NEIL2/MTMR14",
"LYPLA2/ACAA1/OAT/NEDD4/ARG2/DIS3/ACHE/HMOX1/PYGL/TIMP1/QPRT/FAH/UBE2S/ALDOC/SKP1/KYNU/CLU/CAT/NUDT10/PSME3/HSD17B4/STAM/PCYOX1L/CLGN/FBXL18/AGL/ERAP1/FBXO22/TMUB2/GUSB/FUCA1/UBE2L3/LONP1/NUDT11/PSMB10/FIS1/GPX1/NA/SNX17/HGS/ATP2B4/GPC1/FAP/DLD/PON2/WFS1/RBM24/UCHL3/OPTN/RBM38/HSPBP1/GSTM3/ALDH1L2/TGFB1I1/ERLIN2/DFFA/CBS/FOXK1/BCL2/TBK1/AKR1C3/HSD17B11/BAD/FYN/SCARB1/SRPX/CDK4/IDH1/RCN3/EPHX1/NUDT19/STX12/UFM1/ITGB4/CPTP/METTL3/SNX5/NAGLU/HAX1/PRKCA/NEIL2/MTMR14"
), Count = c(10L, 22L, 22L, 33L, 42L, 43L, 43L, 73L, 78L, 84L
)), class = "data.frame", row.names = c(NA, -10L))
Pathway GeneRatio FDR
1 Alpha-amino acid biosynthetic proc. 0.13333333 3.851756
2 Carboxylic acid biosynthetic proc. 0.06686930 4.941747
3 Organic acid biosynthetic proc. 0.06646526 4.941747
4 Small molecule biosynthetic proc. 0.05583756 5.919270
5 Carboxylic acid metabolic proc. 0.04272635 5.207657
6 Oxoacid metabolic proc. 0.04261645 5.207657
GeneID
1 ASNS/CTH/ASS1/PSAT1/CBS/ATP2B4/OAT/PHGDH/MTHFD1/PLOD3
2 ASNS/CTH/ASS1/ACLY/PSAT1/OXSM/CBS/NA/CASP1/ATP2B4/MTHFD1/KYNU/OLAH/AKR1C3/OAT/PHGDH/PLOD3/HSD17B12/FABP5/ERLIN2/PRMT3/CBR1
3 ASNS/CTH/ASS1/ACLY/PSAT1/OXSM/CBS/NA/CASP1/ATP2B4/MTHFD1/KYNU/OLAH/AKR1C3/OAT/PHGDH/PLOD3/HSD17B12/FABP5/ERLIN2/PRMT3/CBR1
4 ASNS/COQ9/COQ5/CTH/COQ6/ASS1/ACLY/PSAT1/OXSM/CBS/FDXR/PC/NA/CASP1/ATP2B4/MTHFD1/KYNU/ERLIN2/OLAH/AKR1C3/OAT/PHGDH/PLOD3/ALDOC/HSD17B12/ADK/FABP5/PTPN2/PRMT3/SIRT5/AACS/ACSS3/CBR1
5 ACAA1/OAT/ASNS/ARG2/QPRT/FAH/ALDOC/KYNU/CTH/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/PC/AKR1C3/NA/DLD/CASP1/ATP2B4/MTHFD1/ALDH1L2/OLAH/FOXK1/ACAD9/GPX1/LYPLA2/AACS/PHGDH/PLOD3/SLC27A1/HSD17B12/FABP5/GFPT1/NUDT19/ERLIN2/PRMT3/GSS/CBR1
6 ACAA1/OAT/ASNS/ARG2/QPRT/FAH/ALDOC/KYNU/CTH/ASS1/ACLY/HSD17B4/PSAT1/IDH1/EPHX1/OXSM/CBS/PC/AKR1C3/NA/DLD/CASP1/ATP2B4/MTHFD1/ALDH1L2/OLAH/FOXK1/ACAD9/GPX1/LYPLA2/AACS/PHGDH/PLOD3/SLC27A1/HSD17B12/FABP5/GFPT1/NUDT19/ERLIN2/PRMT3/GSS/ABHD14B/CBR1
Count
1 10
2 22
3 22
4 33
5 42
6 43
and this is my code:
install.packages("ggplot2")
install.packages("dplyr")
install.packages("ggalluvial") # For Sankey-like plots
install.packages("tidyr") # For unnest function
library(dplyr) # For data manipulation
library(tidyr) # For separate_rows function
data <- read.csv(file = "C:/Users/Sofia/Downloads/data.csv", sep=";")
sankey_dot <- function(data,
sankey.text.size = 3,
sankey.x.axis.text.position = 1,
sankey.x.axis.text.size = 12,
molecule.label = "GeneID",
dot.color = "Count",
dot.position = 6,
dot.label = "Pathway",
dot.text.size = 12,
dot.scale = 0.72,
dot.x = 0.59,
dot.y = -0.173,
dot.width = 0.48,
dot.height = 1.33,
...) {
Data collation after KEGG or GO enrichment analysis
data3 <- data %>%
dplyr::select("GeneID", "Pathway", "FDR", "Count", "FoldEnrichment") %>%
dplyr::distinct() %>%
dplyr::arrange(FDR, descreasing = TRUE)
data3$FoldEnrichment <- data3$FoldEnrichment %>% round(digits = 2)
bubble chart
if (dot.label == "Pathway") {
p1 <- ggplot(
data = data3,
aes(
x = FoldEnrichment,
y = stats::reorder(Pathway, FoldEnrichment)
)
) +
geom_point(aes(size = Count, color = -log10(FDR))) +
scale_size_continuous(range = c(2, 6)) +
theme_bw() +
theme(
axis.text.x = element_text(
size = "Count",
colour = "black", vjust = 1
),
axis.text.y = element_text(
size = "Count",
colour = "black", hjust = 1
)
) +
scale_colour_distiller(palette = dot.color, direction = -1) +
labs(x = "FoldEnrichment", y = "") +
theme_bw() +
theme(
axis.title = element_text(size = 12),
axis.text = element_text(size = 11),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
} else if (dot.label == "GeneID") {
p1 <- ggplot(
data = data3,
aes(
x = FoldEnrichment,
y = stats::reorder(GeneID, FoldEnrichment)
)
) +
geom_point(aes(size = Count, color = -log10(FDR))) +
scale_size_continuous(range = c(2, 6)) +
theme_bw() +
theme(
axis.text.x = element_text(
size = "Count",
colour = "black", vjust = 1
),
axis.text.y = element_text(
size = "Count",
colour = "black", hjust = 1
)
) +
scale_colour_distiller(palette = dot.color, direction = -1) +
labs(x = "FoldEnrichment", y = "") +
theme_bw() +
theme(
axis.title = element_text(size = 12),
axis.text = element_text(size = 11),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
}
data_sankey <- data %>%
dplyr::arrange(FoldEnrichment, descreasing = FALSE) %>%
dplyr::select(herb, molecule_id, target, dot.label) %>%
as.data.frame() %>%
dplyr::distinct()
df <- data_sankey %>% ggsankey::make_long(colnames(data_sankey))
if (dot.label == "Description" && molecule.label == "molecule_id") {
df$node <- factor(df$node, levels = c(
data_sankey$Description %>% unique(),
data_sankey$target %>% unique(),
data_sankey$molecule_id %>% unique(),
data_sankey$herb %>% unique()
))
} else if (dot.lable == "ID" && molecule.lable == "molecule_id") {
df$node <- factor(df$node, levels = c(
data_sankey$ID %>% unique(),
data_sankey$target %>% unique(),
data_sankey$molecule_id %>% unique(),
data_sankey$herb %>% unique()
))
} else if (dot.lable == "Description" && molecule.lable == "molecule") {
df$node <- factor(df$node, levels = c(
data_sankey$Description %>% unique(),
data_sankey$target %>% unique(),
data_sankey$molecule_id %>% unique(),
data_sankey$herb %>% unique()
))
} else if (dot.lable == "ID" && molecule.lable == "molecule") {
df$node <- factor(df$node, levels = c(
data_sankey$ID %>% unique(),
data_sankey$target %>% unique(),
data_sankey$molecule %>% unique(),
data_sankey$herb %>% unique()
))
}
color settings
mycol <- cols4all::c4a("rainbow_wh_rd", length(unique(df$node)))
mycol2 <- sample(mycol, length(mycol))
Sankey diagram
p2 <- ggplot(df, aes(
x = x,
next_x = next_x,
node = node,
next_node = next_node,
fill = node,
label = node
)) +
ggsankey::geom_sankey(
flow.alpha = 0.5,
node.color = NA,
show.legend = FALSE
) +
ggsankey::geom_sankey_text(
size = sankey.text.size,
color = "black",
hjust = 1,
position = position_nudge(x = -0.05)
) +
ggsankey::theme_sankey(base_size = 20, base_family = "sans") +
scale_fill_manual(values = mycol2) +
theme(axis.title = element_blank()) +
theme(axis.text.x = element_text(
size = sankey.x.axis.text.size,
hjust = sankey.x.axis.text.position,
vjust = 10, colour = "black"
))
p3 <- p2 + theme(plot.margin = unit(c(0, dot.position, 0, 0), units = "cm"))
p4 <- cowplot::ggdraw() +
cowplot::draw_plot(p3) +
cowplot::draw_plot(p1,
scale = dot.scale,
x = dot.x, y = dot.y,
width = dot.width,
height = dot.height
)
return(p4)
}
Can anyone help me to have an input like this?
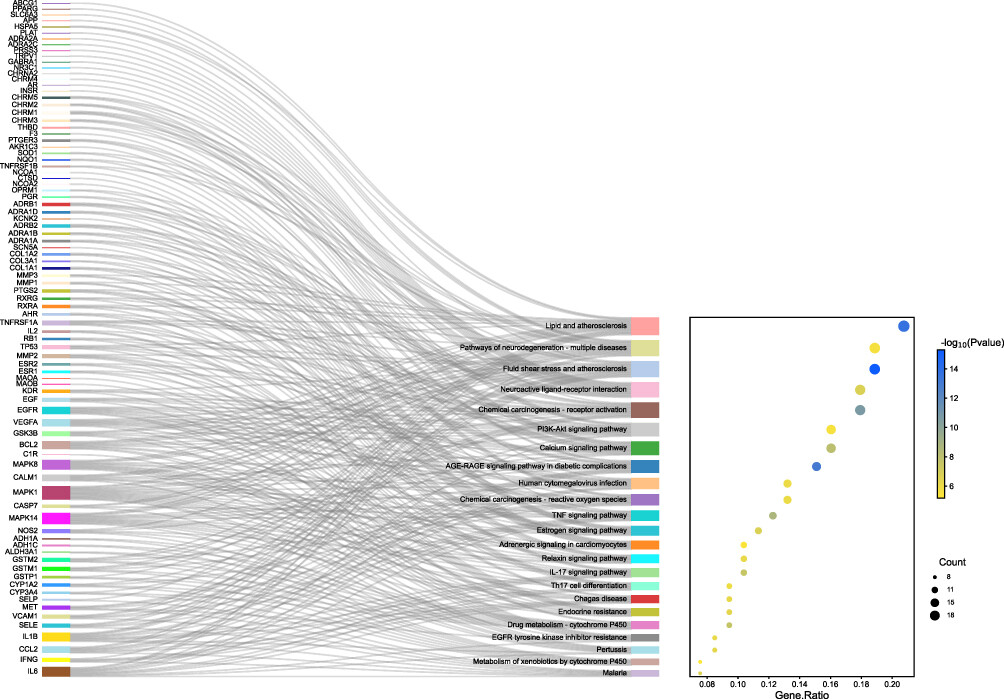
thank you