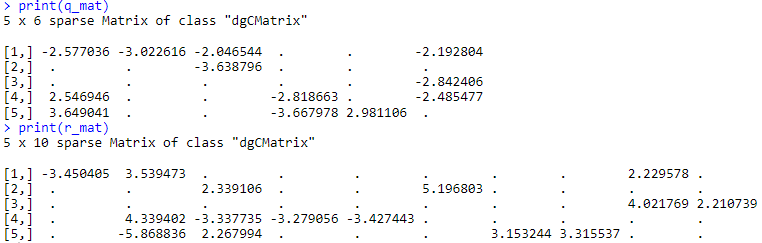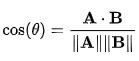Hi Posit Community,
I have two matrices of Z-scores (differential gene expression). They have variables (genes) as rows, samples (observations) as columns:
Query: 1000 rows x 2000 samples
Reference: 1000 rows x 720000 samples
They are sparse as they have been filtered on significance of the Z-scores, setting all values below the threshold to zero.
The overall goal is to calculate cosine similarities between each sample (column) across the two matrices (Query_I to Reference_J) resulting in a 2000 x 720000 matrix, then scaling the cosine similarities based on the number of non-zero overlaps between the i,j samples (as a measure of support for each similarity), turning that into a distance measure and then finally do hierarchical clustering (complete linkage) using these distances.
I am working in an HPC environment where I'm capped at 350GB memory, 32 CPU cores. I have access to GPU clusters as well if necessary.
library(Matrix)
# Simulate some (100x smaller) data
q_mat <- Matrix(data = rnorm(n = 2e4, mean = 0, sd = 0.9, nrow = 1000)
r_mat <- Matrix(data = rnorm(n = 7.2e6, mean = 0, sd = 0.9, nrow = 1000)
# Filter Z-scores on significance level
threshold <- 2
q_mat[abs(q_mat) < threshold] <- 0
r_mat[abs(r_mat) < threshold] <- 0
# Drop samples that do not contain atleast 1 significant value
q_mat <- q_mat[,colSums(abs(q_mat)) != 0]
r_mat <- r_mat[,colSums(abs(r_mat)) != 0]
# My matrices have become sparse: Approximately 98% of values are zero.
q_mat <- as(q_mat, "dgCMatrix")
r_mat <- as(r_mat, "dgCMatrix")
# Cosine similarities can easily be found with
cosine_mat <- qlcMatrix::cosSparse(q_mat, r_mat)
# Boolean matrices which tells us where each matrix is non-zero
q_idx <- q_mat != 0
r_idx <- r_mat != 0
# Getting number of overlaps
n_overlaps <- t(q_idx) %*% r_idx
# Scale the cosines by log2 of the number of overlaps
scaling_factor <- log2(n_overlaps+1)
scaled_cosines <- cosine_mat * scaling_factor
# Turn the cosines into a distance measure
distances <- max(scaled_cosines) - scaled_cosines
# Now turn this into a dist() object somehow for hclust() to use.
First question:
The cosine similarities above will have no contributions from genes where both samples are zero, but it will have contributions from genes where one of the samples is zero and the other is not. As a zero here can both mean below detection limit or truly zero but I have no way of knowing which, they need to be left out from my cosine calculation. In this way, I base the similarity only on genes which are differentially expressed in both.
# The code above will calculate the cosine similarity in the following way:
v1 <- c(0, -3, 0, 0, 5, 3, 4, 0)
v2 <- c(0, 2, 5, 0, 0, -2, 2, 3)
cosine <- v1 %*% v2 / (sqrt(sum(v1^2)) * sqrt(sum(v2^2)))
# What I want is to calculate cosines only from values where both vectors are non-zero:
idx <- v1 != 0 & v2 != 0
v1_nz <- v1[idx]
v2_nz <- v2[idx]
cosine2 <- v1_nz %*% v2_nz / (sqrt(sum(v1_nz^2)) * sqrt(sum(v2_nz^2)))
However, this filtering needs to be done based on overlaps between each set of samples compared. To my understanding, since my matrices are large (and this will be reused for many different query and reference matrices of similarly large sizes) I will want to avoid loops / apply() for subsetting genes and then calculating cosines.
Idea 1: Use Kronecker product to get a boolean matrix for filtering all combinations
kron_qr <- q_idx %x% r_idx
Output would be a sparse matrix along the lines of:
| sampleQ_1:sampleR_1 | sampleQ_1:sampleR_2 | ... | sampleQ_2000:sampleR_720000 | |
|---|---|---|---|---|
| gene1:gene1 | 0 | 1 | ... | 0 |
| gene1:gene2 | 0 | 0 | ... | 0 |
| ... | ... | ... | ... | ... |
| gene1000:gene1000 | 1 | 0 | ... | 0 |
If I'm not entirely wrong this would take up an unfeasible amount of memory. Estimates:
# Dense (float precision):
1000*1000*2000*720000*8 / 2^30 # 10728836 GB
# Sparse (float precision):
(1000*1000*2000*720000*8 / 2^30) * 0.02 # 214577 GB
And that is ignoring any overhead.
However, I am not interested in the rows comparing expression of dissimilar genes (rows), meaning that I wasted computation time and memory on them. Is there any linear algebra voodoo I could invoke to instead get a dim 1000 x (2000*720000) matrix out instead? That would reduce the estimated size for the sparse matrix to ~215GB which may fit in memory, provided there is not too much other overhead.
Idea 2: Tensor
The above could perhaps be translated into a sparse-tensor framework using library(tensorr).
For my final cosine matrix of dim 2000 x 720000 the values each have to be calculated from two vectors of length 1000 containing 1-1000 non-zero values.
Maybe I can create a rank 3 tensor (i,j,k)=(2000 x 720000 x 1000) where the 1000-dimensional part is a boolean vector of "sampleQ_i != 0 & sampleR_j != 0". This boolean vector could then be used to subset the two vectors on which I base the i,j-th cosine similarity.
Second question:
It seems to me that due to the sizes of the matrices in question, the above methods are problematic even if I can request access to more memory. Maybe I am overcomplicating it?
Is my best bet then to do some sort of parallelized apply() ? I'm a bit worried about the runtime, and have not used parallelization approaches with R before. How would I approach this?
Sorry for the long post, and thank you for your time!